The Pharma Legal Handbook: Italy




 Join industry executives in staying informed on pharma regulations in Italy.
Join industry executives in staying informed on pharma regulations in Italy.
Regulation, Pricing, Clinical Trials, Marketing, Manufacturing, Trademarks, Patents, and more!
Get your pharmaceutical legal and regulatory questions answered in The Pharma Legal Handbook – a must-have guide for any company operating in the country or looking to enter the market.
Prepared in association with DLA Piper, a leading international law firm.
If legal handbook content is updated, you will receive a free updated PDF for up to one year after purchase.
January 2020
1. Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs: Italy
The key facts about regulatory reforms in Italy. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
Cannabinoid Drugs
1. Are Cannabinoid Drugs authorized in your country?
In Italy, the use of recreational cannabis is prohibited under Presidential Decree no. 309/1990 (the “Presidential Decree”).
However, it is possible to use cannabis for medical purposes.
Indeed, the Decree of the Ministry of Health no. 98 of 28 April 2007 (the “2007 Decree”) officially recognized and acknowledged the therapeutic properties of cannabis and updated the tables annexed to the Presidential Decree listing active principles and their medicinal compositions (in particular, Delta-9-tetrahydrocannabinol (THC), Trans-delta-9-tetrahydrocannabinol (Dronabinol), and Nabilone were added to the table II-B of the Presidential Decree and admitted to importation).
The 2007 Decree also authorized physicians in Italy to prescribe extemporaneous preparations (preparazioni magistrali) made by pharmacists using either Dronabinol or a plant-based active substance based on Cannabis for medical use.
The Decree of the Ministry of Health of 23 January 2013 (the “2013 Decree”) has subsequently inserted additional active compounds of cannabis origin in the table II-B of Presidential Decree.
In addition to the national legislation, there are also a number of regional laws allowing the administration and, in some cases, the production of medicine and extemporaneous preparations based on cannabinoids for therapeutic purposes. These regional laws provide for easier and cheaper access to cannabinoid-based drugs or have authorized new clinical trials or production of these drugs.
2. What are the regulatory authorities with jurisdiction over Cannabinoid Drugs?
According to art. 2.1, letter d) of the Presidential Decree, the Regulatory authority with jurisdiction over cannabinoid drugs is the Ministry of Health (“MoH”) which also represents the State Cannabis Body (Organismo statale per la cannabis), as provided under the Convention on Narcotic Drugs adopted in New York on 30 March 1961.
3. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Cannabinoid Drugs?
Cannabinoid drugs are subject to the general provisions set out by the Presidential Decree for the authorization, use, import, export and market of narcotic drugs and psychotropic substances.
In particular, pursuant to art. 17 of Presidential Decree, anyone who intends to carry out any of the aforementioned activities shall obtain the prior authorization from the MoH, and another, additional, permit is requested to import or export such products. The authorizations issued by the MoH have a two-years duration and are subject to the payment of a government concession fee. However, the obligation to obtain the aforementioned authorizations does not apply to pharmacies with reference to the purchase, sale and administration of narcotic drugs and psychotropic substances.
As for the pricing and reimbursement, there is not a specific legal framework for cannabinoid drugs. Indeed, the general framework provided for by the Decree of the Ministry of Health of 2 August 2019 (the “2019 Decree”) applies to cannabinoid drugs as well.
4. Which are the cannabinoid drugs that have received market approval to date?
As of today, to our knowledge, only one medicinal product based on extracts of cannabis sativa has been authorised for marketing (i.e., SativexR), which is generally prescribed to alleviate the symptoms in adult patients suffering from moderate to severe spasticity due to multiple sclerosis, who have not shown an adequate response to other, different, antispasmodic drugs and have shown a clinically significant improvement with reference to spasticity symptoms during an initial test period of therapy.
5. Who can prescribe Cannabinoid Drugs?
Since 2007 Decree came into force, any physician is eligible to prescribe cannabis products and synthetic cannabinoids for therapeutic use; subsequently, the 2013 Decree also authorized physicians to prescribe cannabis-based drugs of vegetable origins.
6. Is there a list of doctors authorized to prescribe Cannabinoid Drugs?
There is no list of physicians authorized to prescribe cannabinoid drugs.
7. What approvals or notifications are required to prescribe Cannabinoid Drugs?
No specific approval or notification is required to prescribe cannabinoid drugs.
There are, however, some general procedural obligations set forth by articles 38 to 45 of the Presidential Decree to be followed.
In addition, art. 61 of Presidential Decree requires pharmacists to keep a specific register to record the quantities and qualities of drugs included in the first four tables of the Presidential Decree that have been sold or used during the year.
8. Which organizations are authorized to sell/distribute Cannabinoid Drugs available?
According to art. 17 of the Presidential Decree, anyone who intends to produce, employ, import, export, receive for transit, trade in any capacity or otherwise possess for trade narcotic and psychotropic substances referred to in art. 14 of the Presidential Decree (which includes THC and its derivatives) has to obtain the prior authorization from the Ministry.
The market authorization for SativexR is held by GW Pharma. However, since SativexR is considered a “class H” drug, it is not dispensable in pharmacy, but it can be distributed by the Local Health Companies, A.S.L and Hospital Health Companies, A.S.O..
9. Is there a list of retailers/ distributors authorized to sell Cannabinoid Drugs?
Yes, according to art. 16 of the Presidential Decree, the list of all companies and entities authorized to grow, produce, use and market narcotic and psychotropic substances is kept by the MoH, published on the Official Gazette and annually updated through decree of the MoH.
10. Are there proposals for reform or significant change to the regulation of Cannabinoid Drugs?
N/A.
11. When are they likely to come into force?
N/A.
Medicinal Cannabis
12. Is Medicinal Cannabis authorized in the country?
Yes, medicinal Cannabis is legal in Italy since the approval and entry into force of the 2007 Decree, which officially recognized the therapeutic properties of Cannabis and updated the tables annexed to the Presidential Decree listing active principles and their medicinal compositions.
However, the Decree of the Ministry of Health of 9 November 2015 (“2015 Decree”) provides that the medical use of cannabis cannot be considered a therapy on its own, but rather a treatment supporting standard therapies, when the latter have not produced the desired benefits, or have resulted in side effects that cannot be tolerated, or require dosage increases that could lead to the appearance of side effects.
The 2015 Decree also established the uses for which medical cannabis may be prescribed.
13. What are the regulatory authorities with jurisdiction over Medicinal Cannabis?
According to art. 2.1, letter d) of the Presidential Decree, the Regulatory authority with jurisdiction over cannabinoid drugs is the Ministry, which also represents the State Cannabis Body (Organismo statale per la cannabis), as provided under the Convention on Narcotic Drugs adopted in New York on 30 March 1961.
14. What is the regulatory framework for the authorization, pricing, and reimbursement of Medicinal Cannabis?
Article 17 and 19 of the Presidential Decree provide the authorization procedure to be followed for growing, producing, importing, exporting, commercializing and deploying medical cannabis. Such authorization requirement, however, does not apply to pharmacies.
The procedure for setting the price of medical cannabis is set out in point 6 of the Technical Annex to the 2015 Decree, which states that the price of medical cannabis is to be fixed by revising the Annex A to the Ministerial Decree of 18 August 1993 every two years by means of Decree of the Ministry. The current price is set to 9 Euros/gram.
As for the reimbursement, the 2015 Decree, in point 3 of the relevant Technical Annex establishes that the rembursement of medical cannabis by the National Healthcare System (“NHS”) is subject to the indications issued by the single Regions or Autonomous Provinces. As of today, 8 Regions authorized the reimbursement for all the uses of medical Cannabis established by the aforementioned 2015 Decree and one (Apulia) even extended the uses for which medical cannabis can be prescribed and refunded.
15. How is the production and import of Medicinal Cannabis regulated and by which agencies/authorities?
According to art. 17 of the Presidential Decree, anyone who intends to grow, produce, import, export, commercialize and deploy medical cannabis shall obtain the prior authorization from the MoH. However, there is no need for such authorization in case of pharmacies, which are expressly exempted from the authorization process.
Note that if someone has already obtained the authorization from the Ministry and intends to import or export medical cannabis, an additional permit from the Ministry shall be obtained and the procedural obligations established by articles 51 to 55 of the Presidential Decree shall be complied with.
The authorization issued by the Ministry has a two-years duration and is subject to the government concession fee. Additionally, art. 3 of the 2015 Decree states that the MoH (i) has decisional power over the areas in which Cannabis plants can be cultivated (ii) establishes the imported, exported and distributed quantities on the national territory, (iii) authorises the import, export, wholesale distribution and store of Cannabis plants and plant material, and (iv) provides for the determination of the production quotas of active substance of plant origin based on Cannabis on the basis of the requests of the Regions and Autonomous Provinces.
16. What approval or notifications are necessary to produce or import Medicinal Cannabis?
According to art. 1 of the 2015 Decree, the MoH has the power to import medical cannabis and to authorise its import, according to the procedure established by articles 17 and 27 of the Presidential Decree. Such procedure prescribes the obtainment of a first, prior authorization for the production of the drugs included in the tables attached to the Presidential Decree and, in case of import, a second permit is needed from the MoH.
17. What is the regulatory framework for the marketing and distribution of Medicinal Cannabis?
The regulatory framework for the marketing and distribution of medicinal cannabis is established by articles 37 to 42 of the Presidential Decree, under which anyone who intends to market and distribute narcotic drugs and psychotropic substances has to present a formal request to the MoH, providing the information required by art. 37 of the Presidential Decree.
Subsequently, the MoH, after carrying out the necessary verification procedure, may grant the authorization and eventually set the requirements and conditions to be complied with. Additional procedural requirements are set forth by articles 38 to 42 of the Presidential Decree.
18. How can patients obtain Medicinal Cannabis?
According to the 2015 Decree, patients can be prescribed medical cannabis from physicians if they present one of the following symptoms (indicated in point 4.1 of the Technical Annex to the 2015 Decree):
• analgesia in pathologies involving spasticity, associated with pain resistant to conventional therapies;
• analgesia in chronic pain where treatment with non-steroidal anti-inflammatory drugs or with cortisone or opioid drugs has proved ineffective;
• the anticinetosic and antiemetic effect in nausea and vomiting, caused by chemotherapy, radiation therapy, HIV therapies, which cannot be achieved with traditional treatments;
• the stimulating effect of appetite in cachexia, anorexia, loss of appetite in patients with cancer or AIDS and in anorexia nervosa, which cannot be obtained by standard treatment;
• the hypotensive effect in treatment-resistant glaucoma;
reduction of involuntary body and facial movements in Gilles de la Tourette syndrome which cannot be obtained with standard treatments.
19. Who can prescribe Medicinal Cannabis?
Since the entry into force of the 2013 Decree, which amended the Presidential Decree and inserted cannabis-based active compounds of plant origin in the table II-B, physicians are authorized to prescribe medicinal cannabis.
20. Is there a list of doctors authorized to prescribe Medicinal Cannabis?
N/A.
21. What approvals or notifications are required to prescribe Medicinal Cannabis?
No specific approval or notification is required to prescribe medicinal cannabis. However, there are particular requirements that the relevant prescription sheet has to comply with in order to be valid, as established in art. 43 of the Presidential Decree. In addition, art. 61 of Presidential Decree requires pharmacists to keep a specific register to record the quantities and qualities of drugs included in the first four tables of the Presidential Decree that have been sold or used during the year.
22. Where is Medicinal Cannabis available?
Medicinal cannabis is available at pharmacies, in accordance with the Presidential Decree.
23. Is there a list of retailers authorized to sell Medicinal Cannabis?
On 4 April 2022, the MoH and the Ministry of Defence signed the Collaboration Agreement for the launch of a pilot project for selecting companies able to cultivate cannabis plants to be supplied to the Military Chemical and Pharmaceutical Plant in Florence for the manufacture of medicines and pharmaceutical raw materials (i.e., Cannabis FM2). However, pending the full availability of Cannabis FM2 produced by Florence military facility, the Ministry confirmed on its website that it will continue to authorize the import of cannabis for the preparation of extemporaneous preparations..
24. Are there proposals for reform or significant change to the regulation of Medicinal Cannabis?
N/A. It is worth noting, however, that a proposal to hold a popular referendum for the liberalization of cannabis cultivation has been recently rejected by the Constitutional Court with sentence no. 51 of 2022.
Opioid Drugs
25. Are Opioid Drugs authorized in your country?
According to the Presidential Decree, opium and opioid drugs are authorized in Italy and listed in the Table I attached to the Presidential Decree. In addition to that, Law no. 38 of 15 March 2010 has amended art. 14 of the Presidential Decree in order to simplify the access procedure for medicines used in pain therapy, by providing the possibility of also prescribing the drugs included in the Table section A of the Presidential Decree.
26. What are the regulatory authorities with jurisdiction over Opioid Drugs?
According to art. 2.1, letter d) of the Presidential Decree, the regulatory Authority for opioid drugs is the Ministry.
27. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Opioid Drugs?
Opioid drugs are subject to the same provisions as other narcotic and psychotropic substances, so there is not a specific legal framework for their authorization and the procedure provided for by article 17 of the Presidential Decree applies.
As for the pricing and reimbursement, such aspects are negotiated between the Italian Drug Agency (“AIFA”) and the pharmaceutical companies, as provided by the 2019 Decree, which establishes AIFA jurisdiction with regard to the negotiation procedures related to the price and reimbursement of medical products to be paid by the NHS.
28. Which are the Opioid drugs that have received market approval to date?
Based on the list of medicinal products that received the necessary market authorization from AIFA, the approved Opioid Drugs include, inter alia, the following active principles:
• Fentanyl;
• Paracetamol;
• Codeine;
• Naloxone.
29. Who can prescribe Opioid Drugs?
All physicians in Italy can prescribe opioid drugs.
30. Is there a list of doctors authorized to prescribe Opioid Drugs?
N/A.
31. What approvals or notifications are required to prescribe Opioid Drugs?
No specific notification or authorization is required to prescribe opioid drugs. Furthermore, Law no. 38 of 15 March 2010, concerning provisions to ensure access to palliative care and pain therapy, has amended the Presidential Decree in order to simplify the access procedure for medicines used in pain therapy, by providing the possibility of prescribing also the drugs included in the Table section A of the Presidential Decree. In addition to that, in case the opioid drugs are used within a pain therapy, there is a general obligation to keep track of the levels of pain suffered by the patient, in order to assess the suitability of the therapy deployed.
In addition, art. 61 of Presidential Decree requires pharmacists to keep a specific register to record the quantities and qualities of drugs included in the first four tables of the Presidential Decree that have been sold or used during the year.
32. Which organizations are authorized to sell/distribute Opioid Drugs available?
According to art. 17 of the Presidential Decree, anyone who intends to deliver narcotic and psychotropic substances referred to in art. 14 of the Presidential Decree has to obtain the prior authorization from the Ministry. However, such obligation does not apply to pharmacies.
33. Is there a list of retailers/ distributors authorized to sell Opioid Drugs?
Yes, according to art. 16 of the Presidential Decree, the list of all companies and entities authorized to grow, produce, use and market narcotic and psychotropic substances is kept by the MoH, published on the Official Gazette and annually updated by decree of the same MoH.
34. Are there proposals for reform or significant change to the regulation of Opioid Drugs?
N/A.
35. When are they likely to come into force?
N/A.
Also from this Legal Handbook
2. Regulatory Reforms: Italy
The key facts about regulatory reforms in Italy. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
1. Are there proposals for reform or significant change to the healthcare system?
The most recent changes in legislation relate to the clinical trial sector, where Legislative Decree No. 52/2019 and the Decree of the MoH dated November 30, 2021, which was published in the Italian Official Gazette on February 16, 2022, have introduced new rules according to which data and results obtained from non-profit clinical trials funded by the Italian NHS can be assigned to third parties for registration and marketing purposes provided that either the sponsor or the assignee refunds and reimburses any direct or indirect associated costs (please see Chapter 2).
Also, on June 26, 2022 Law No. 62/2022 on ‘Transparency rules on relationships between manufacturing companies, healthcare professionals and organizations’ (“Sunshine Act”) entered into force. With said law, the Italian legislators has introduced, for the first time, disclosure and transparency obligations concerning relationships of economic relevance between companies manufacturing medicinal products, instruments, equipment, goods and services (even of non-health nature) (“Manufacturing Companies”), and healthcare professionals and organizations.
According to the Sunshine Act, reporting and disclosure obligations apply, among other things, to (i) agreements and disbursements of money, goods, services or other benefits performed by a Manufacturing Company in favor of healthcare professionals and/or organizations; and (ii) agreements between Manufacturing Companies and healthcare professionals and/or organizations providing direct or indirect benefits for the latter (participation in conferences, training events, committees, etc.). Law No. 62/2022 introduces pecuniary sanctions for the nondisclosure and the late disclosure of data as well as for the submission of incomplete information.
The Sunshine Act also provides for a public register that will be established on the institutional website of the Ministry of Health within six months from the entry into force of the law (i.e. by December 26, 2022). Said register will be used to publish data submitted by Manufacturing Companies and those relating to sanctions, which will be accessible for a period of five years.
2. When are they likely to come into force?
Directory of local institutions
Also from this Legal Handbook
3. Patents & Trademarks: Italy
All legal aspects surrounding patents & trademarks in Italian Pharma. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
1. What are the basic requirements to obtain patent and trademark protection?
The regulation of Italian patents and trademarks is mainly contained in the Industrial Property Code (Legislative Decree No. 30/2005, hereinafter “I.P.C.”). Some provisions on patents and trademarks are also contained in the Italian Civil Code.
As regards the patent for invention, which includes the pharmaceutical patent, inventions from any field of technology which are new, involve an inventive activity and are capable of industrial application are patentable (art. 45(1) of the I.P.C.). The requirements for patentability of the invention are: novelty, inventive step, industrial application and lawfulness (Articles 46 to 50 of the I.P.C). Specific rules are provided for certain types of patents, including special rules on the biotechnological patent.
With respect to trademarks, these must first comply with the provisions of Article 7 of the I.P.C., establishing which “signs” can be registered as trademarks. The requirements for registration are: novelty, distinctiveness and lawfulness (Articles 12 to 14 of the I.P.C.).
2. What agencies or bodies regulate patents and trademarks?
The regulation of Italian patents and trademarks is contained in the Italian legislation, which also transposes some EU Directives on the matter. The Italian Patent and Trademark Office, which is part of the Ministry of Economic Development, is the registration office for both patents and trademarks.
3. What products, substances, and processes can be protected by patents or trademarks and what types cannot be protected?
As regards patents, inventions in any field of technology that are new, involve an inventive step, and are capable of industrial application may be patented.
According to article 45 of the I.P.C., if considered “as such”, the following are not regarded as inventions:
• discoveries, scientific theories and mathematical methods;
• schemes, rules and methods for performing mental acts, playing games or doing business and computer programs;
• presentations of information.
Article 45 above also provides that (i) methods for surgical or therapeutic treatment of the human or animal body and methods of diagnosis applied to the human or animal body; and (ii) plant varieties and animal breeds and essentially biological processes for the production of animals or plants, cannot be patented.
With regard to trademarks, pursuant to Article 7 of the I.P.C, all signs, in particular words, including names of persons, drawings, letters, numerals, sounds, the shape of the product or its packaging, color combinations or shades, can be registered as trademarks, provided that they are capable of:
• distinguishing the goods or services of an enterprise from those of other enterprises; and
• being represented in the register in such a way as to enable the competent authorities and the public to clearly and accurately determine the subject matter of the protection granted to the relevant holder.
Limitations on the possibility of registering a trademark are set out in Article 8 on portraits of persons, names, notorious signs, Article 9 on shape marks, and Article 10 on badges, emblems and escutcheons of the I.P.C.
4. How can patents and trademarks be revoked?
Patents may be declared null and void if the invention lacks the requirements for patentability and in the other cases provided for by Article 76 of the I.P.C. Patents may also expire for non-payment of fees (Art. 75 of the I.P.C.).
Trademarks may be declared null and void if the invention lacks one of the requirements set forth in Article 7 and in the other cases provided for in Article 25 of the I.P.C. Forfeiture is also provided for in cases of vulgarization, supervening unlawfulness and non-use (Article 26 of the I.P.C.).
The bodies responsible for performing the relevant checks and declaring the nullity / forfeiture are the Italian Patent and Trademark Office and the Italian judicial authority.
5. Are foreign patents and trademarks recognized and under what circumstances?
Registration titles have territorial validity on the basis of the national registration office chosen. Thus, unless European or international titles include Italy, only Italian patents and trademarks are recognized in Italy.
However, certain rights are given to unregistered but actually used trademarks and a right of priority in case of filing in a Country which is a party to an international convention ratified by Italy that recognizes the right of priority (Art. 4 f the oI.P.C.).
6. Are there any non-patent/trademark barriers to competition to protect medicines or devices?
The recipes, list of ingredients, components, way of manufacturing and any other business information relevant to the medicine / device can be handled by the owner as a trade secret and protected as such when the following conditions apply: (i) the information is not generally known or easily accessible by others; (ii) it has an economic value inasmuch as it is confidential; (iii) it is subject to reasonable measures to preserve its confidentiality.
Obviously, trade secrets are not a suitable tool when reverse engineering allows third parties to replicate the finding.
7. Are there restrictions on the types of medicines or devices that can be granted patent and trademark protection?
No restrictions, save for what indicated above at Question 6 (iii)
8. Must a patent or trademark license agreement with a foreign licensor be approved or accepted by any government or regulatory body?
No approval or acceptance is needed, but licenses must be recorder with the Italian Patents and Trademarks Office in order to be enforceable against third parties. Note that licenses on applications that have not yet been published will not be possible until publication.
Also from this Legal Handbook
4. Product Liability: Italy
The low-down on the situation regarding product liability in Italy. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
1. What types of liability are recognized in your jurisdiction?
The Italian regulatory framework, with reference to the liability for defective products, consists of:
• Contractual liability, governed by article 1218 of the Italian Civil Code;
• Liability for defective products as established by article 1490 of the Italian Civil Code;
• Liability for lack of quality, governed by article 1497 of the Italian Civil Code;
• Liability for defective products regarding sale agreements concluded by customers established by articles 114 seq. of Legislative Decree 206/2005 (Consumer Code);
• Tort liability as foreseen by article 2043 of the Italian Civil Code.
It is understood that, depending on the actual circumstances of the case, criminal liabilities may arise for the individual(s) responsible of the relevant facts (e.g. injuries, death).
2. How do these types of liabilities apply to the manufacturers of medicines and devices?
The relevant framework is established by the Consumer Code, as ruled by the Supreme Court of Cassation in various judgments concerning the liability of manufacturers of medicinal products for damages caused to users due to defects of said products (e.g., Court of Cassation – 10/05/2021, No. 12225). As for the liability, pursuant to art. 120 of the Consumer Code, the injured user — in order to prove the liability of the manufacturer — must prove the defect (which can be a material defect, a design defect, or a defect on the information about the product and its correct use); the damage suffered; and the causal connection between the two. The manufacturer is instead required to prove the facts that may exclude his liability (e.g., force majeure). Product liability is therefore presumed, and not strict, since it does not depend on the establishment of the manufacturer’s culpability, but rather on the proof of the existence of a defect in the product. As for the definition of “defective product,” the security threshold, below which the product is to be regarded as defective, arises when the product does not offer the safety that may legitimately be expected, having regard to circumstances such as the manner and the period in which the product was put into circulation, its presentation, its obvious characteristics, the instructions and warnings given, the use for which the product may reasonably be intended, and the conduct that may reasonably be expected in connection therewith.
Furthermore, the Pharma Code also sets a specific regulatory framework for manufacturers of medicines and medical products. In particular, the Pharma Code sets specific obligations for manufacturers of the latter: In order to access the market, manufacturers need to obtain a prior authorization issued by AIFA. To obtain such authorization, manufacturers have to ensure compliance with various provisions, including, but not limited to, conformity with principles and guidelines on good manufacturing practice for medicinal products; establishment, implementation and maintenance of an effective pharmaceutical quality system; establishment of a system that registers and examines complaints; information obligations toward AIFA with reference to every defect that could give rise to the recall of the product or supply restrictions. Manufacturers may be held liable to the extent the breach of such duties is linked to the injuries suffered by the user; and AIFA, pursuant to art. 146 of the Pharma Code, may recall or suspend the market authorization and/or the production of the relevant product.
3. Does potential liability extend to the manufacturer only or could claims extend to corporate executives, employees, and representatives?
The Italian legal system is based upon the principle of personal liability, which also applies to legal entities that have been recognized with legal personality, so that, as a general rule, the legal entity should be the only one held liable with reference to the manufacturing of defective products.
It is also possible for the supplier to be held responsible, in case the manufacturer has not been identified and the supplier has failed to inform the injured party of the identity and domicile of the former.
In case of criminal ramifications, the liability would primarily be held by the individuals (e.g. representatives of the corporate operator) in charge of the relevant activities, while the manufacturer could remain liable for corporate responsibility under Legislative Decree No. 231/2001.
Furthermore, Law No. 24/2017 has reformed the discipline of the liability of healthcare operators both when they operate as freelancers and when they operate in any capacity within facilities. In such cases, liability is related to the negligence of the healthcare provider and may be contractual or non-contractual in nature. There may therefore be a concurrence of responsibility between the manufacturer and the healthcare provider if the damage suffered by the victim depends both on a defect in the product and on the negligent conduct of the healthcare operator.
4. How can a liability claim be brought?
A liability claim can be brought upon judicial or administrative courts by any interested party, that is, any consumer that suffered damages linked to the defective product. This matter was further clarified by the Italian Supreme Court of Cassation in judgment No. 13458 of May 29, 2013, in which it has been established that “all persons who have in some way been exposed, even occasionally, to the risk deriving from the defective product are entitled to take action; the protection granted to the ‘user’ has to be interpreted in a broad sense and, therefore, undoubtedly to a natural person – as is made clear by the identification of the damage that can be compensated in that ‘caused by death or personal injury’ and by the limitation of the material damage that can be compensated – but not exclusively to the ‘consumer’ or non-professional user.”
It is also possible for the damaged consumer to appeal for the resolution of disputes at the “Directorate-General for the Market, Competition, Consumer Protection and Technical Regulations” (so-called Direzione generale per il mercato, la concorrenza, la tutela del consumatore e la normativa tecnica) of the Ministry of Economic Development.
5. What defenses are available?
The defendant can base its defense upon any defense that is foreseen by the Civil Code or the Consumer Code, for example, that it complied with the relevant obligations or that the damage was related to exceptional circumstances, like force majeure. Moreover, article 118 of the Consumer Code enumerates specific situations in which manufacturer liability is to be excluded, specifically:
a) If the manufacturer did not put the product into circulation;
b) If the defect that caused the damage did not exist when the manufacturer put the product into circulation;
c) If the manufacturer did not manufacture the product for sale or for any other form of distribution for consideration, nor did he manufacture or distribute it in the exercise of his professional activity;
d) If the defect is due to the conformity of the product with a mandatory rule of law or a binding measure;
e) If the state of scientific and technical knowledge at the time when the manufacturer put the product into circulation did not yet permit the product to be regarded as defective;
f) With reference to the manufacturer or supplier of a component part or raw material, if the defect is wholly due to the design of the product into which the part or raw material was incorporated or it was due to the conformity of the latter with the instructions given by the manufacturer who used it.
Lastly, under the Consumer Code, it would not be possible for the manufacturer to ground its defense on the presence of a contractual clause that excludes or limits its liability toward the injured consumer, as this type of clauses are to be considered null and void vis-à-vis consumers.
Needless to say, all possible defensive strategy and defensive arguments are to be identified on a case-by-case basis, in light of actual circumstances and based on the role held and actions taken by each person, including the damaged consumer, the healthcare professionals, any operator within the supply chain, and/or any individual within the manufacturer’s organization.
Also from this Legal Handbook
5. Traditional Medicines and OTC Products: Italy
The ins and outs of traditional medicines and OTC products in Italy. Prepared in association with DLA Piper, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for USD 99.
1. What are the regulatory requirements for traditional, herbal, complementary, or alternative medicines and devices?
a. Medicinal products
To obtain a marketing authorization for medicinal products through a national, mutual recognition or decentralized procedure, the applicant is always required to submit an application in compliance with Art. 8 of the Pharma Code (which also apply to centralized procedures pursuant to Art. 6 of Regulation (EC) No. 726/2004) which provides the list of mandatory documents and information for the relevant regulatory assessment, except that, in case of generic drugs, it is possible to submit a simplified or bibliographic application under Art. 10 and 11 of the Pharma Code, for biosimilar drugs, the application shall comply with the provisions of Art. 10, paragraph 7, of the Pharma Code , whereas, in case of medicinal products containing active substances used in the composition of authorized medicinal products but not hitherto used in combination for therapeutic purposes, Art. 12 of the Pharma Code provides that the scientific evaluation by the AIFA shall refer to the results of new pre-clinical tests or new clinical trials relating to that combination and that it shall not be necessary to provide scientific references relating to each individual active substance.
Pursuant to Art. 21 and following of the Pharma Code, herbal medicinal products which fulfil certain criteria can benefit from a simplified registration procedure (“traditional-use registration”) providing for a reduction in the documents and information that the applicant must submit to the AIFA, compared with those requested for the registration of traditional medicines. In case the application for a traditional-use registrations refers to herbal substances, preparations and combinations thereof included in the list provided for by Art. 16f of Directive 2001/83/EC, further reductions in terms of documents and information apply.
b. Medical devices
According to the MDR, a medical device can be placed on the market or put into service only if it complies with the provision of the MDR when duly supplied and properly installed, maintained and used in accordance with its intended purpose. To this end, a medical device must meet the general safety and performance requirements set out in the MDR applicable to it, taking into account its intended purpose. Demonstration of conformity with the general safety and performance requirements includes a clinical evaluation.
Applicable safety and performance requirements depend on the risk class of the medical device which is also of relevance for the purposes of identifying the specific procedure to be followed for the affixing of the “CE marking”, that is a marking by which the manufacturer indicates that the device is in conformity with the applicable requirements set out in the MDR and which is precondition for placing the same device on the EU market. Classifications rules for medical devices are set out in Annex VIII to the MDR. The application of said criteria leads to four main classes: I (lowest risk), IIa, IIb, and III (highest risk). In addition, Class I devices can be further subdivided into Is – sterile condition, Im – measuring function and Ir – reusable surgical.
While for low-risk devices the relevant manufacturer declares the conformity of its products by issuing the EU declaration of conformity and affixes the CE marking, for high-risk medical device the demonstration of conformity with the general safety and performance requirements necessary to affix the CE marking requires the involvement of a notified body (i.e. a conformity assessment body designated in accordance with the MDR).
With specific regard to certificates of conformity issued by the notified bodies, they are valid for the period indicate therein, which cannot exceed five years. On application by the manufacturer, the validity of the certificate may be extended for further periods, each not exceeding five years, based on a re-assessment in accordance with the applicable conformity assessment procedures. Any supplement to a certificate remains valid as long as the certificate which it supplements is valid.
2. Can these traditional, herbal, complementary, or alternative products be advertised directly to the public?
Please refer to the answer to Question 17 in Chapter 3 which apply to all categories of medicinal products.
3. What health, advertising, and marketing claims may be made for traditional, herbal, complementary, or alternative products?
There are no specific legal provisions with regard to the type of language to be used in the advertising of medicines to HCPs and to the general public, for example, regarding the use of expert terminology. For those categories of medicinal products whose advertising to the general public is not prohibited and that need to be authorized, the claims must be approved by the MoH.
Having said that, it should be noted that Art. 114 of the Pharma Code lays down the general principle according to which the advertising of a medicinal product must encourage the rational use of the medicinal product, by presenting it objectively and without exaggerating its properties and must not be misleading. Further rules and restrictions apply to the advertising of medicinal products to the general public. Indeed, pursuant to Art. 116 of the Pharma Code, this form of advertising must:
• be set out in such a way that it is clear that the message is an advertisement and that the product is clearly identified as a medicinal product;
• include the following minimum information:
– the name of the medicinal product, as well as the common name if the medicinal product contains only one active substance;
– the information necessary for correct use of the medicinal product; and
-an express, legible invitation to read carefully the instructions on the package leaflet or on the outer packaging, as the case may be.
Art. 117 of the Pharma Code set forth certain limitations on the content of advertising messages addressed to the general public by establishing that they cannot include any material which, amongst other things:
• gives the impression that a medical consultation or surgical operation is unnecessary, in particular by offering a diagnosis or by suggesting treatment by mail;
• suggests that the effects of taking the medicine are guaranteed, are unaccompanied by adverse reactions or are better than, or equivalent to, those of another treatment or medicinal product;
• suggests that the health of the subject can be enhanced by taking the medicine;
• suggests that the health of the subject could be affected by not taking the medicine;
• is directed exclusively or principally at children;
• refers to a recommendation by scientists, health professionals or persons who are neither of the foregoing but who, because of their celebrity, could encourage the consumption of medicinal products;
• refers, in improper, alarming or misleading terms, to claims of recovery.
Lastly, with specific regard to herbal medicinal product Art. 27(4) of the Pharma Code provides that any advertisement must contain the following statement: “Traditional herbal medicinal product for use in specified indication(s) exclusively based upon long-standing use”.
4. What are the regulatory requirements for over-the-counter (non-prescription) medications?
Please refer to the answer to Question 1 which also apply to over-the-counter medicinal products.
5. Are there any limitations on locations or channels through which OTC products may be sold?
In Italy, OTC products can be sold in pharmacies, para-pharmacies and retail businesses set up in Municipalities with more than 12,500 inhabitants. Moreover, since 2015, prescription-free medicinal products, including OTC products, can also be sold online by those pharmacies, para-pharmacies and retail businesses which have obtained an ad hoc prior authorization from the local competent health authority pursuant to Art. 112-quarter of the Pharma Code.
6. What health, advertising, and marketing claims may be made for OTC products?
The rules and limitations on health, advertising, and marketing claims of traditional medicines also apply to OTC products.
7. Can OTC products be marketed or advertised directly to the public?
OTC products can be advertised to the general public provided that the relevant advertising message is previously authorized by the MoH.
8. What is the mechanism by which a prescription-only product can be converted to an OTC product?
A prescription-only product can be converted to an OTC product either on the AIFA own initiative or upon request of the relevant MAH; in the latter case, the conversion application must be expressly approved by the AIFA.
9. What are the requirements for the importation of either traditional medicines or OTC products?
Importing and placing on Italian market medicines from extra-EU countries requires a manufacturing authorization from the AIFA. Moreover, the Decree of the MoH dated February 11, 1997 governs the import in the Italian territory of medicines authorized abroad.
Also from this Legal Handbook
6. Marketing, Manufacturing, Packaging & Labeling, Advertising: Italy
All about marketing, manufacturing, packaging & labeling, advertising in Italy. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
1. What is the authorization process for the marketing of new drugs, biologics, medical devices, over the-counter medications, and other medicinal products?
Please refer to the answer to Question 2 in Chapter 1.
2. What is the authorization process for the marketing of generic versions of these products?
Please refer to the answer to Question 6 in Chapter 1.
3. What are the typical fees for marketing approval?
Pursuant to art. 158 of the Pharma Code, applicable fees for marketing approval are established by a decree of the MoH after having heard the AIFA and are automatically updated on the basis of the Italian National Statistical Institute (“ISTAT”) of the cost of living. Updated fee are published on the AIFA’s institutional website. Applicable fees depend on several factors, including the type of medicinal product for which the marketing authorization is requested, the relevant strength, pharmaceutical form, etc., and may range from few to several tens of thousands of Euros.
4. What is the period of authorization and the renewal process?
Please refer to the answer to Question 5 in Chapter 1.
5. What are the requirements, if any, for post-approval pharmacovigilance?
Under the Pharma Code, the MAH must ensure a system to collect and maintain a record of suspected adverse events and equally ensure a safety reporting system to the AIFA. Specifically, the MAH shall set up a risk management system for each medicine, appoint a qualified person who shall be responsible for pharmacovigilance activities and notify his or her name to the AIFA. The pharmacovigilance activities may lead to the suspension or revocation of product approvals by the AIFA, which ensure compliance with the provisions of law inspecting the MAH premises, records and documents in relation to the pharmacovigilance activities covered. Moreover, according to the Pharma Code the MAH shall appoint a responsible person for distribution and a scientific service, which must be independent from the marketing department.
AIFA is competent for pharmacovigilance compliance and, to this end, has the power to perform inspections at the marketing authorization holders’ premises and access to their records and related documents.
6. Are foreign marketing authorizations recognized?
Foreign marketing authorizations are not recognized in Italy. It should be noted, however, that pursuant to the Decree of the MoH dated 11 February 1997 the same MoH may authorize a physician to import a medicine duly licensed in a country outside of Italy necessary to treat a patient if such an import is justified on the basis of objective exceptional criteria and provided that the relevant medicinal product is used in compliance with Italian law and in accordance with conditions of use authorized in the country of origin. Moreover, in case of shortage of medicine at a national level, the MoH can authorize the import of the same medicine available outside of Italy.
7. Are parallel imports of medicines or devices allowed?
Yes. With specific regard to medicinal products, the parallel import is subject to the AIFA’s prior authorization.
8. What are the restrictions on marketing practices such as gifts, sponsorships, consultancy agreements, travel and entertainment, or other incentives for healthcare organizations and individual medical practitioners?
a. Gifts
According to Art. 123 of the Pharma Code on advertising of pharmaceutical products (the principles of which also apply to medical devices), no gifts, pecuniary advantages or benefits in kind can be supplied, offered or promised to healthcare professionals (“HCPs”) with the exception of those of “negligible value” and that, in any case, are related to the activity performed by the relevant HCP / pharmacist. In this respect, the Guidelines on the Scientific Information adopted by the Conference of Regions and Autonomous Provinces fix the “negligible value” in a maximum annual amount of EUR 20.00 for pharmaceutical company for each HCP / pharmacist.
Furthermore, pursuant to Art. 170 of the Consolidated Text of Health Laws (Royal Decree No. 1265 of 27 July 1934 as amended), the HCP who inappropriately receives money or other benefits to inflate the number of drug prescriptions or other medical products for pharmaceutical use is punishable with the imprisonment of up to one year and with a pecuniary fine up to EUR 520.
If pecuniary advantages are offered to HCPs, the general rules regarding bribery would apply.
Additional rules on gifts to HCPs are provided for in the Code of Conducts adopted by Farmindustria (the Italian Association of Pharmaceutical Companies) and Confindustria Dispostivi Medici (the Italian Association of Medical Device Companies).
b. Sponsorships, travel and entertainment
Pursuant to Art. 124 of the Pharma Code, pharmaceutical companies may support congresses and events on topics related to the use of medicinal products provided that they obtain the AIFA’s prior authorization. Art. 124 above also provides that pharmaceutical companies may contribute to costs related to the participation of qualified HCPs in said congresses and event with respect to travel and accommodations, with the exclusion of accompanying persons. In this respect, hospitality cannot be extended to a period of time exceeding 12 hours prior to the beginning and/or following the end of the congress / event. Moreover, the characteristics of the hospitality must not prevail over the technical-scientific purposes of the event.
Further rules and restrictions on sponsorships, travel and entertainment are established in the Codes of Conduct adopted by Farmindustria (the Italian Association of Pharmaceutical Companies) and Confindustria Dispostivi Medici (the Italian Association of Medical Device Companies). By way of example, the Farmindustria’s Code of Conduct provides that, in case of rail transport, all travel classes are permitted with the exception of the Executive class, whereas, in case of flights, these must be exclusively in economy class. Exceptions apply with respect of international flights with a duration exceeding 6 hours, in which case it is possible to provide business class tickets only for speakers and moderators included in the official agenda of the event. All events must be held in appropriate venues that are conducive to the main purpose of the event, avoiding those that are known for their entertainment facilities or are ‘extravagant’. The organization or sponsoring of events taking place, or the provision of hospitality to participants, in resorts, vessels, manors located outside of the urban area, to farmhouses, golf clubs and spa facilities is not allowed. Moreover, seaside locations cannot be selected as a venue for an event from June 1 to September 30. The same prohibition applies to mountain locations from December 1 to March 31, as well as from July 1 to August 31.
c. Consultancy agreements
Under Italian law, consultancy agreements between pharmaceutical and medical device companies and HCPs are allowed. It should be noted, however, that pursuant to Art. 53 of Legislative Decree No. 165/2001, HCPs who are employed by a public hospital or entity cannot be entrusted with the performance of paid services, including consultancy agreements, unless they have obtained the prior authorization from the public hospital / entity they work for. In case the prescribed authorization is obtained, the beneficiary of the services must communicate to the relevant public hospital / entity the amount of the compensation paid to the HCP within 15 days from the payment date. In case of violation of the provisions of Art. 53 above, private persons are sanctioned with a pecuniary fine equal to twice the amount paid to the public employee.
Lastly, Legislative Decree No. 165/2001 prohibits former public employees (including HCPs formerly employed by a public hospital or entity) who, in the last three years of their employment relationship, have exercised authoritative or negotiating powers on behalf of a public administration from performing, during the three years following the termination of their employment relationship, any professional or work activity for the benefit of private persons in respect of whom such powers have been exercised. Agreements entered into in violation of the above-mentioned prohibition are null and void and private persons entering into said agreements shall be prohibited from negotiating with public administrations for the following three years and must return any possible compensation received in connection with the same agreements.
9. How is the manufacturing of medicines and devices regulated and by which agencies?
a. Medicinal products
Pursuant to Art. 50 of the Pharma Code, the manufacturing of medicinal products is subject to a prior authorization (“Manufacturing License”). The Manufacturing License is granted by the AIFA after having carried out an inspection aimed at verifying that the applicant possess, amongst other things, (i) suitable and sufficient premises, technical equipment and control facilities for the production and storage of medicines, and (ii) a so-called Qualified Person responsible for verifying that each batch of medicinal products is manufactured and checked in compliance with the applicable legislation and in accordance with the requirements of the MA and, with respect to medicinal products coming from non-EU countries, that each production batch has undergone a full qualitative analysis, a quantitative analysis of at least all the active substances and all the other tests or checks necessary to ensure the quality of medicinal products in accordance with the requirements of the MA. The Qualified Person must be qualified and reliable. Qualification must be substantiated by (i) holding a license to practice as a chemist, pharmacist or physician, have a university diploma in the relevant field, (ii) at least two years of practical experience in the field, and (iii) being licensed to exercise his/her profession and enrolled with the relevant professional register. Manufacturing Licenses are normally granted within 90 days for the filing of a complete application
b. Medical devices
The production of medical devices does not require a manufacturing license. However, when placing their devices on the market or putting them into service, manufacturers must ensure that they have been designed and manufactured in accordance with the requirements of the MDR and/or IVDR, as applicable.
10. Are local manufacturing requirements compatible with Good Manufacturing Practices (GMPs) as defined by the U.S. Food & Drug Administration and/or the European Medicines Agency?
Yes. Under the Pharma Code, manufactures of medicinal products must comply with the principles and guidelines of good manufacturing practice for medicinal products as laid down by the same Pharma Code as well as with the other directives issued at the EU on this matter.
11. What is the inspection regime for manufacturing facilities?
a. Medicinal products
Under the Pharma Codes, the AIFA is responsible for ensuring that all legislative provisions on medicinal products are complied with. To this end, the AIFA carries out period inspections, including unannounced inspections, on production plants and other premises where medicines and active substances are manufactured, imported, controlled and stored and has the power to take samples of medicinal products and to review and take copy of all relevant documents. In performing these activities, the AIFA may request the support of the National Institute of Health, the National Institute for Occupational Safety and Prevention, Local Health Authorities and of other public bodies.
Moreover, pursuant to Art. 71 of the Pharma Code, manufacturers are required to carry out continued self-inspections in order to check the application of and compliance with Good Manufacturing Practices and to adopt the necessary corrective actions or preventive measures. Self-inspections, as well as all subsequent corrective actions, must be recorded and the relevant records must be kept for a period of at least 10 years.
b. Medical devices
In Italy, the MoH is the national authority responsible for enforcing the MDR and the IVDR and can perform periodic inspections, require manufacturers to provide justifications, and implement corrective measures.
In particular, Art. 93 of the MDR entrusts the competent authorities of the EU member States (in Italy, the MoH), with the task of performing appropriate checks on the conformity characteristics and performance of devices including, where appropriate, a review of documentation and physical or laboratory checks on the basis of adequate samples. To this end, the MoH may carry out both announced and, if necessary, unannounced inspections of the premises of economic operators and has the power to confiscate, destroy or otherwise render inoperable devices that present an unacceptable risk where it deems it necessary to do so in the interests of the protection of public health.
12. Are manufacturing facilities open for inspection by foreign inspectors or third-party inspectors as authorized by the FDA/EMA?
In Italy, manufacturing facilities are normally inspected by the AIFA, which is the national supervisory authority for manufacturing and is responsible for verifying, also on behalf of the EU, that manufacturers satisfy the requirements concerning manufacturing laid down in the Italian and EU legislation.
That said, it is worth mentioning that, pursuant to Art. 19 of Regulation (EC) No. 726/2004, in case the European Commission is informed of serious differences of opinion between Member States as to whether a manufacturer established within the EU satisfies the requirements concerning manufacturing, the Commission may, after consultation with the Member States concerned, request an inspector from the supervisory authority to undertake a new inspection of the manufacturer. In such a case, the inspector in question is accompanied by two inspectors from Member States which are not party to the dispute or by two experts nominated by the Committee for Medicinal Products for Human Use.
13. What are the requirements for storage, packaging, and handling of medicines and devices and their constituent components?
The requirements for storage, packaging, and handling of medicines and devices vary depending on the concerned drugs. In any case, whoever stores medicines must obtain a prior authorization from the Italian Regional Authority of the place where the warehouse is located.
14. What information must be included in medicine and device labeling?
a. Medicinal products
For medicinal products, the information to be reported in the labelling are identified in Art. 73 of the Pharma Code. These include, the name of the medicinal product followed by the strength and the pharmaceutical form; a statement of the active substances expressed qualitatively and quantitatively per dosage unit or according to the form of administration for a given volume or weight, using their common names; the pharmaceutical form and the contents by weight, by volume or by number of doses of the product; the method of administration and, if necessary, the route of administration; a special warning that the medicinal product must be stored out of the reach and sight of children; the expiry date in clear terms (month/year); special storage precautions, if any; the name and address of the marketing authorization holder; the number of the authorization for placing the medicinal product on the market; the manufacturer’s batch number; the price of the medicinal product and whether it is reimbursed by the NHS.
Specific rules apply to the labelling of herbal medicinal products, medicinal products containing radionuclides and homeopathic medicinal products.
b. Medical devices
According to the MDR, the label shall bear, amongst other things, the following particulars: the name or trade name of the device; the details strictly necessary for a user to identify the device, the contents of the packaging and, where it is not obvious for the user, the intended purpose of the device; the name, registered trade name or registered trade mark of the manufacturer and the address of its registered place of business; where applicable, an indication that the device contains or incorporates a medicinal substance, including a human blood or plasma derivative, or tissues or cells, or their derivatives, of human origin, or tissues or cells of animal origin, or their derivatives; the lot number or the serial number of the device; the Unique Device Identification carrier; the time limit for using or implanting the device safely; an indication of any special storage and/or handling condition that applies; warnings or precautions to be taken that need to be brought to the immediate attention of the user of the device, and to any other person; where applicable, an indication that the device is intended for single use and whether it has been reprocessed; if the device is custom-made, the words “custom-made device”; and for active implantable devices, the serial number, and for other implantable devices, the serial number or the lot number.
15. What additional information may be included in labeling and packaging?
a. Medicinal products
Under Art. 73 of the Pharma Code, in addition to mandatory information, the label may include, upon notification to the AIFA, the name of the person that, based on a specific agreement with the relevant MAH, actually markets the medicinal product throughout the Italian territory (i.e. the exclusive distributor / concessionaire).
b. Medical devices
Under Italian law, there no specific provisions on this point.
16. What items may not be included in labeling and packaging?
a. Medicinal products
Under Italian law, there no specific provisions on this point.
b. Medical devices
Pursuant to Art. 7 of the MDR, neither the labelling nor the instructions for use may include text, names, trademarks, pictures and figurative or other signs that may mislead the user or the patient with regard to the device’s intended purpose, safety and performance by ascribing functions and properties to the device which the device does not have; create a false impression regarding treatment or diagnosis, functions or properties which the device does not have; suggest uses for the device other than those stated to form part of the intended purpose for which the conformity assessment was carried out.
17. What are the restrictions and requirements for the marketing and advertising of medicines and devices?
a. Marketing
As a general rule, only duly authorized medicinal products and medicinal devices bearing the CE marking can be marketed in Italy.
Exceptions to this rule apply in very limited cases. With respect to medicinal products, the most remarkable example of these exceptions are “compassionate use programs” which provide for the use of unauthorized medicines that are expected to help patients with life-threatening, long-lasting or seriously debilitating illnesses, which cannot be treated satisfactorily with currently authorized medicines . Compassionate use programs are coordinated and implemented by Member States, which set their own rules and procedures. In Italy, the Decree of the Ministry of Health dated 7 September 2017 allows the compassionate use of medicines for (1) patients suffering from rare or life-threatening diseases, if no other valid therapeutic alternative is available; (2) patients who have already been treated with clinical benefit in a similar clinical trial; or (3) patients who cannot be included in a clinical trial .
Similarly, in cases of exceptional need and urgency, in the interest of a single patient, the MoH can authorize the compassionate use of medical devices bearing no mark because the conformity assessment has not been conducted or completed. Compassionate use requires MoH authorization and the approval of the relevant Ethics Committee.
b. Advertising and promotion
Regulatory framework
The advertising and promotion of medicines and medical devices is governed by national legislation, as well as by regional legislation (i.e., laws and regulations issued by the various Italian regions) and industry codes.
In detail, the following pieces of legislation apply to the advertising of medicines and medical devices in Italy:
a) the Pharma Code;
b) Legislative Decree No. 46/1997, implementing Directive 93/42/EEC on medical device; and
c) The Decree of the MoH dated 23 February 2006 on medical devices advertising.
Specific instructions on advertising of medicines and medical devices through different communication channels, including the internet, SMS/MMS and social networks, are established by various guidelines issued by the MoH.
At a regional level, the promotion of medicines and medical devices to HCPs is regulated by the “Guidelines for regional regulations regarding scientific information relevant on medicines” dated 26 April 2006, as well as by the guidelines issued by each Italian Region.
Lastly, additional rules on advertising of both medicines and medical devices are also set out in the codes of conduct adopted by national and international industry associations, such as the Farmindustria and the EFPIA Codes for medicines, and the Confindustria Dispositivi Medici and the MedTech Europe Codes for medical devices. Said codes only apply to pharma and medical devices companies that are members of such industry associations.
Advertising to the general public
Advertising to the general public of prescription medicines or medicines containing psychotropic or narcotic substances is forbidden. The prohibition on advertising to the general public also applies to medicines that are reimbursed, even partially, by the NHS. The distribution of medicines to the public for promotional purposes is also prohibited.
In publications, radio or television broadcasting, or any non-promotional messages to the general public, it is prohibited to mention the name of a medicinal product in a context where this results in the promotion of the consumption of the product.
Similarly, it is forbidden to advertise to the general public custom-made medical devices and medical devices that must be ordered by, chosen by or ultimately used with the assistance of, an HCP.
The advertising of medicines and medical devices that do not fall within one of the above-mentioned categories is subject to prior authorization by the MoH. This authorization is implicitly granted if the MoH does not raise any objection within 45 days from the date of the filing of the relevant application.
Promotion to HCPs
Broadly speaking, the promotion of medicines and medical devices to HCPs is governed by concerns for accuracy in information, adequacy in education and training of medical sales representatives, and the avoidance of undue influence over HCPs’ decision-making processes (including purchasing decisions by public entities). To this end, there are a number of restrictions on how information can be presented to HCPs with respect to seminars, panel discussions, promotional events and the like.
Any advertising of medicines to persons qualified to prescribe or supply such products shall always include the summary of the product characteristics authorized at the time the advertising is disseminated, specify the supply classification of the same product and the sale price and the conditions for its reimbursement by the NHS, if any.
The documentation on the medicinal product must be filed with the AIFA and can be supplied to the HCP after the expiry of a term of 10 days from the filing.
18. Where can medicines and devices be sold or delivered? Can medicines and devices be sold or delivered via post?
In Italy, medicinal products can be delivered to patients by pharmacies, hospitals or similar healthcare structures. The applicable supply regime is established by the AIFA’s Technical-Scientific Committee taking into consideration the issues relating to the safe use of the relevant medicinal products.
As a general rule, medicinal products subject to medical prescription (i.e. Class A, Class H and certain Class C medicines) can only be provided to patients by territorial and hospital pharmacies. Pursuant to Art. 32 of Law Decree 201/2011, Class C medicinal products not subject to medicinal prescription can also be sold by para-pharmacies and retail businesses set up in Municipalities with more than 12,500 inhabitants. Moreover, since 2015, prescription-free medicinal products, including over-the-counter products, can also be sold online. It should be noted, however, that the online sale of prescription-free medicinal products can only be performed by those pharmacies, para-pharmacies and retail businesses which have obtained an ad hoc prior authorization from the local competent health authority.
With respect to medical devices, the existing legislation does not specifically regulate the relevant sales channel, with the consequence that all medical devices, including those whose use requires the assistance of HCPs, can be sold by pharmacies, para-pharmacies, orthopaedic stores and even directly by the relevant manufacturers. Moreover, Art. 6(1) of the MDR provides that medical devices can be offered online by information society services provided that the same devices complies with the provisions of the same MDR. As neither the MDR nor the Italian legislation currently regulate or limit usable online sales channels, it follows that medical devices can be offered online through e-commerce sites belonging to manufacturers, distributors or importers as well as market places. Also, the online sale of medical devices does not require any authorization and can be performed both with respect to legal and natural persons, without any distinction between HCPs and the general public. It should be noted, however, that the above is without prejudice to the rules and restrictions applicable to the advertising of medicinal devices to the general public.
19. What are the restrictions and requirements for electronic marketing and advertising via email, by Internet, social media, and other channels?
a. Electronic marketing
As regards medicines, only prescription free medicinal products can be sold online and only by pharmacies, para-pharmacies and retail businesses which have obtained an ad hoc prior authorization from the local competent health authority. For medical devices, there are no similar restrictions or authorization requirements.
b. Electronic advertising
Advertising to the general public
Under Italian law, it is prohibited to advertise to the general public prescription-only medicines, drugs containing psychotropic or narcotic substances and medicines reimbursed, even partially, by the NHS as well as custom-made medical devices and medical devices that must be ordered by, chosen by or ultimately used with the assistance of, an HCP, whereas, for all other products, the dissemination of advertising messages to the general public is subject to the prior authorization of the MoH.
Without prejudice to the foregoing, specific instructions on advertising of medicines and medical devices to the general public through different communication channels, including emails, the internet and social networks are established by various guidelines issued by the MoH (the “Guidelines”). According to the Guidelines, it is possible to disseminate advertising messages via email provided that, at the time of the submission of the relevant request for authorization, the applicant company declares that the relevant messages will only be sent after having obtained the consumer’s consent and that the latter may freely revoke at any time his/her consent and request the cancellation of his/her data. Similarly, in case of advertising messages disseminated through websites, the applicant must include in the relevant request for authorization the web address where the advertisement will be published. Also, the concerned company must clearly identify in the same websites the advertising message(s) and include and include a message specifying that the MoH’s authorization only refers to the same message(s).
Stricter rules apply to the dissemination of adverting messages through Social Networks as the use of these channels is regarded suspiciously by the MoH. Indeed, as clarified by the same MoH in its Guidelines, the advertising message authorized by the MoH has a static nature, meaning that it cannot be modified neither by the holder of the relevant product nor by any other person. Considering that the use of Social Networks, which allow users to express their opinions, to advertise medical devices compromises said static nature, the MoH resolved to allow the advertising of medical devices through Social Networks only with respect to certain hosting providers (i.e. Facebook, YouTube and Instagram) and to subject the same advertising to specific conditions and limitations identified in the Guidelines.
Promotion to HCPs
The dissemination of information about medicine and medical devices to HCPs via email and/or the internet is permitted without the MoH’s authorization. However, with specific regard to the internet, the spread of this information must be on websites or on web pages only intended to be viewed by HCPs. To this end, companies must inform the users through a specific disclaimer that the information provided on the sites are only addressed to HCPs.
20. May medicines and devices be advertised or sold directly to consumers?
Please refer to the answers to Question 17, 18 and 19 above.
21. How is compliance monitored?
Compliance with rules governing the adverting of medicines and medical devices is monitored by the MoH with the support of the Anti-adulteration and Health Units of the Carabinieri Corp.
22. What are the potential penalties for noncompliance?
Companies that violate the provisions of Italian law regarding the promotion of medical devices are punished with administrative fines up to €15,493.71. In addition, the MoH can order the immediate cessation of the advertising and require the infringing party to rectify or clarify the advertising in accordance with the instructions provided by the same MoH.
In the case of misleading advertising, the Italian Antitrust Authority may order the infringing party to stop any illegal activity, require the infringing party to rectify the relevant statements and publish them at the latter’s expenses, and issue administrative fines up to €500,000.
As for pharmaceutical products, in case of any violation of the rules on advertising to the general public, Article 118 of the Pharma Code provides that the MoH can order the immediate cessation of the advertising and the dissemination, at the expense of the infringing party, of a corrective statement in accordance with the instructions provided by the MoH, unless the MoH decides to proceed by its own initiative. The company is also subject to a pecuniary fine ranging up to €15,600, pursuant to Article 148 of the Pharma Code.
In accordance with Articles 127 and 148 of the Pharma Code, the same sanctions apply in case of breach of the requirements set forth for the advertising to persons qualified to prescribe or supply medicinal products. For products that are reimbursed by the NHS, the violation can also be punished with the suspension of the medicinal product from the reimbursement regime for a period ranging from 10 days to two years. The suspension is adopted after the notification of the violation to the MAH that has the right to file its observations within 15 days from the notification.
Also from this Legal Handbook
7. Preclinical and Clinical Trial Requirements: Italy
Key legal info on preclinical and clinical trial requirements in Italy. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
1. Are clinical trials required to be conducted locally as a condition (stated or implicit) for marketing approval?
No. Under Legislative Decree No. 211/2003 on the application of good clinical practice in the conduct of clinical trials of medicines, clinical trials may be conducted in Italy and in other Member States of the EU. Moreover, according to Regulation (EU) No. 536/2014 on clinical trials on medicinal products for human use, also data generated in clinical trials carried out outside the EU can be used for registration purposed provided that they have been conducted in accordance with principles equivalent to those established in the same Regulation as regards the rights and safety of the subject and the reliability and robustness of the data generated.
2. How are clinical trials funded?
In Italy, clinical trials are classified into:
• “for profit clinical trials”, i.e. clinical trials conducted for registration and marketing purposes or, in any case, for the economic exploitation of the data generated in the trial. For profit clinical trials are sponsored by the pharmaceutical industry; and
• “non-profit clinical trials”, i.e. clinical trials funded by the NHS and conducted by public or non-profit entities which are not the MAH of the relevant investigational drug and do not share any economic interest, also by way of intellectual property rights, with the MAH. Although the pharmaceutical industry may economically support the conduct of non-profit clinical trials, also by providing free of charge products necessary to conduct the trial, the same trials must be carried out to improve clinical knowledge and practice rather than for registration and marketing purposes.
3. What are the requirements for preclinical and clinical trial protocols? Who must approve the protocols?
Trial protocols must describe the objective, design, methodology, statistical considerations, purpose and organization of the clinical trial. Protocols must comply with the requirements set forth in Paragraph D of Annex I to Regulation (EU) No. 536/2014 on clinical trials on medicinal products for human use. Under Italian law, trials protocols must receive the prior approval by the Ethics Committee of each clinical site involved in the trial. Less stringent rules apply to observational studies for which, depending of the specific characteristics of the relevant study, a mere notification of the study protocol to the competent Ethics Committee may be sufficient.
4. What are the requirements for consent by participants in clinical trials?
Pursuant to Legislative Decree No. 211/2003, a clinical trial can only be initiated if, amongst other things, the trial subject or, when the person is not able to give informed consent, his / her legal representative has given his written consent after being informed of the nature, significance, implications and risks of the clinical trial. If the individual is unable to write, oral consent in the presence of at least one witness may be given in exceptional cases.
In order to validly express his consent, the subject or his / her legal representative must be provided with specific information enabling the latter to understand:
• the nature, objectives, benefits, implications, risks and inconveniences of the clinical trial;
• the subject’s rights and guarantees regarding his or her protection, in particular his or her right to refuse to participate and the right to withdraw from the clinical trial at any time without any resulting detriment and without having to provide any justification;
• the conditions under which the clinical trial is to be conducted, including the expected duration of the subject’s participation in the clinical trial; and
• the possible treatment alternatives, including the follow-up measures if the participation of the subject in the clinical trial is discontinued.
The above information must be kept comprehensive, concise, clear, relevant, and understandable to a layperson and must be provided in a prior interview with a qualified member of the investigating team.
Legislative Decree No. 211/2003 and Regulation (EU) No. 536/2014 further provide specific rules on informed consent for cluster trials, emergency situations and trials on minors or incapacitated subjects.
The adequacy and completeness of the written information to be provided to the trial subject or his / her legal representative as well as the procedure aimed at obtaining his / her informed consent must be assessed by the competent Ethics Committee before the trial is approved.
5. May participants in clinical trials be compensated?
Regulation (EU) No. 536/2014 provides that no incentives or financial inducements can be given to the trial subjects or their legally designated representatives, except for compensation for expenses and loss of earnings directly related to the participation in the clinical trial. Such compensation can be given to minors and incapacitated subjects, including their legally designated representatives, and to pregnant or breastfeeding women. In this respect, the MoH has clarified that reimbursements of expenses directly incurred by the subjects for the participation in the clinical trial, such as board and lodging expenses, are not considered as incentives or financial inducements and that said expenses can also be reimbursed to accompanying persons in case of subjects who cannot move independently.
The MoH has also specified that requests for compensations, and related justifications, must in any case be assessed and approved by the competent Ethics Committee and that said compensations must not be used to indemnify subjects for the violation of their rights and safety nor to unduly influence their behaviours.
6. How are participants in clinical trials protected and indemnified against any harm that arises as a result of participation in the trial?
Art. 3 of Regulation EU No. 536/2014 lays down the general principle according to which a clinical trial may be conducted only if, amongst other things, the rights, safety, dignity and well-being of subjects are protected and prevail over all other interests.
In this respect, Regulation EU No. 536/2014 includes several provisions aimed at protecting the safety of trial subjects, such as those requiring the competent authority of the Member State responsible for authorizing the clinical trial to assess the relevant application also on the basis of the risks and inconveniences for the subject, taking account of the safety measures, including provisions for risk minimization measures, monitoring, safety reporting, and the safety plan proposed by the sponsor; those requesting the sponsor to promptly report to the “Eudravigilance” database all suspected unexpected serious adverse reactions occurred during the trial and to file an annual report on the safety of each investigational medicinal product used in the clinical trial; report those requiring the sponsor to adequately monitor the conduct of the trial to verify that the rights, safety and well-being of subjects are protected and to take all appropriate urgent safety measures to protect the subjects where an unexpected event is likely to seriously affect the benefit-risk balance.
At the Italian level, Legislative Decree No. 211/2003 provides that in case there are information casting doubts on the safety of the clinical trial, the MoH may suspend or prohibit the continuation of the same trial.
In terms of indemnification, Art. 76 of Regulation EU No. 536/2014 requires Member States to ensure that systems for compensation for any damage suffered by a subject resulting from participation in a clinical trial are in place in the form of insurance, a guarantee, or a similar arrangement that is equivalent as regards its purpose and which is appropriate to the nature and the extent of the risk. In Italy, Legislative Decree No. 211/2003 expressly provides that a clinical trial may only be commenced if, amongst other things, the sponsor has taken out an insurance policy covering the investigator and sponsor’s liability for any damage to subjects resulting from the participation in the clinical trial. The minimum requirements for said insurance policies are established in the Decree of the MoH dated July 14, 2009. According to the AIFA resolution dated 20 March 2008, observational studies on medicinal products are exempted form insurance policy obligations.
Also from this Legal Handbook
8. Regulatory, Pricing and Reimbursement Overview: Italy
Regulatory, pricing and reimbursement overview in Italy – a legal guide. Prepared in association with Baker Mckenzie, a leading law firm in Italy, this is an extract from The Pharma Legal Handbook: Italy, available to purchase here for GB 119.
1. What are the regulatory authorities with jurisdiction over drugs, biological, and medical devices in your country?
In Italy, the regulatory authorities with jurisdiction over drugs, biologicals, and medical devices are:
• the Ministry of Health (“MoH”), which generally governs the Italian healthcare system and has a more specific role in relation to the regulatory framework concerning medical devices; and
• the Italian Medicine Agency (“AIFA”), a public entity under the supervision of the MoH, which is responsible, amongst other things, for the manufacturing, distribution, import, promotion, advertising, pricing and reimbursement of medicines, including biological drugs.
2. What is the regulatory framework for the authorization, pricing, and reimbursement of drugs, biological, and medical devices?
a.Medicinal products
As regards medicinal products, including biological drugs, the relevant legislative and regulatory framework mainly consists of:
• Regulation (EC) No. 726/2004 laying down EU procedures for the authorization and supervision of medicinal products;
• Legislative Decree No. 219/2006 (the “Pharma Code”) implementing Directive 2001/83/EC relating to medicinal products for human use;
• Law No. 326/2003 with entrusts the AIFA with the task of negotiating the cost of medicines reimbursed by the National Healthcare System (“NHS”) and, more generally, monitoring prices of medicinal products;
• Decree of the MoH dated August 2, 2019 setting forth the criteria and methods according to which the AIFA establishes, through negotiation, the prices of medicinal products reimbursed by the NHS; and
• Law Decree No. 87/2005 governing the price of medicinal products not reimbursed by the NHS.
b.Medical Devices
With respect to medicinal devices, the relevant legislative and regulatory framework mainly consists of:
• Regulation (EU) No. 2017/745 on medical devices and active implantable medical devices (“MDR”);
• Regulation (EU) No. 2017/746 on in vitro diagnostic medical devices (“IVDR”);
• Legislative Decree No. 46/1997, implementing Directive 93/42/EEC on medical device;
• Legislative Decree No. 507/1992, implementing Directive 90/385/EEC on active implantable medical devices; and
• Legislative Decree No. 332/2000, implementing Directive 98/79/EC on in vitro diagnostic medical devices.
In Italy, there are no specific provisions of law governing the pricing and reimbursement of medical devices. Rather, medical devices are directly purchased by the NHS in the context of tender procedures governed by Italian public procurement laws. These laws set forth specific rules governing the award of public supply contracts based on the principles of equal treatment, transparency and free competition and the performance of same pursuantto ad hoc public procurement rules.
Contracting authorities award the relevant supply contract either on the basis of the lower price criteria (option more frequently used for low/medium-technology products) or the most economically advantageous offer (option more frequently used for medical devices characterized by a higher technological level). Exceptions to said rules may apply with respect to medical devices that can only be supplied by one economic operator due to technical reasons or IP rights protection, for which contracting authorities can allow one-to-one negotiated procedures (e.g. in case of urgency).
3. What are the steps to obtain authorization to develop, test, and market a product?
a. Medicinal products
In Italy the development and testing of medicinal products is mainly governed by Legislative Decree No. 211/2003 and Legislative Decree No. 200/2007 on clinical trials on medicinal products for human use according to which trials are subject to the prior authorization issued by the AIFA and the positive opinion of the Ethics Committee(s) relating to the clinical site(s) where same trials are conducted. The above-mentioned Legislative Decrees also set forth specific rules on good clinical practice, informed consent, responsibilities of sponsors and investigators and other entities involved in the clinical trial (e.g., contract research organizations). Less stringent rules apply to observational studies, according to the AIFA’s Resolution dated 20 March 2008.
As to the authorization to market products, under Italian law the approval pathway for new drug applications does not differ from that ordinarily envisaged for all medicines, according to the Pharma Code. That said, simplified procedure for the authorization of generic drugs as well as schemes for expedited approval and programs to encourage the development of new drugs are available.
Under Italian law, there are two main routes for authorizing medicines: a centralized route, governed by Regulation (EC) No. 726/2004 (i.e. the centralized authorization procedure), and a national route, regulated by the Pharma Code, which in turn comprises three different authorization procedures, namely the national procedure, the mutual recognition procedure and the decentralized procedure.
Pursuant to the centralized authorization procedure, pharmaceutical companies submit a single marketing-authorization application to the European Medicines Agency (the “EMA”). The advantage of this procedure is to allow the marketing-authorization holder to market the medicine and make it available to patients and healthcare professionals throughout the EU on the basis of a single marketing authorization. Today, the great majority of new, innovative medicines pass through the centralized authorization procedure in order to be marketed in the EU .
In the EU, marketing authorization applications for biotechnology-derived medicinal products, including biosimilars , are by law reviewed centrally by the EMA. The European Commission issues the decisions concerning the authorization of these medicinal products on the basis of the scientific opinions from the EMA. The resulting marketing authorization is valid in all EU Member States. Other biological medicinal products can also be authorized through the national, mutual recognition or decentralized procedure on the basis of the same scientific and regulatory standards required under the centralized procedure before the EMA.
With specific regard to biosimilars, developers are required to demonstrate through comprehensive comparability studies with the reference biological medicine that:
• their biological medicine is highly similar to the reference medicine, notwithstanding natural variability inherent to all biological medicines;
• there are no clinically meaningful differences between the biosimilar and the reference medicine in terms of safety, quality, and efficacy.
Biosimilar development relies heavily on comparability studies to establish similarity to the reference product. This involves a comprehensive head-to-head comparison of the biosimilar and the reference medicine.
b. Medical devices
To obtain the CE marking for a medical device, the manufacturer must demonstrate that the device complies with the safety and performance requirements laid down in the MDR under the normal conditions of its intended use. To this end, manufactures must provide adequate clinical evidence that, in most cases, are gathered through the conduct of clinical investigations. From May 26, 2021, clinical investigations on medical devices are governed by the MDR according to which ingestions are subject to the prior authorization issued by the competent authority of the Member State(s) in which the same investigation is to be conducted (in Italy, the MoH) and the positive opinion of the Ethics Committee(s) of the concerned clinical site(s).
No prior authorization is required for the marketing of medical devices. However, medical devices can only be marked if they have the CE Marking, that is a marking that the relevant manufacturer affixes on the device to confirm its compliance with the safety and performance requirements set out in the MDR or IVDR, as applicable, and other related EU rules.
Medical devices are classified in four main classes: I (lowest risk), IIa, IIb, and III (highest risk) according to a risk-based approach that takes into account the vulnerability of the human body and the potential risks associated with the devices. This approach uses a set of criteria that can be combined in various ways in order to determine classification, such as duration of contact with the body, degree of invasiveness, local vs. systemic effect, potential toxicity, the part of the body affected by the use of the device and if the device depends on a source of energy.
Determining the risk class of a medical device is essential in specifying the steps required for CE Marking, especially in terms of the choice of conformity assessment procedure and clinical requirements. Indeed, the assessment of the conformity of a device for CE Marking varies according to the risk class and specific features of the respective device. The intervention of a notified body is needed for all high-risk class devices, for which there is also a clinical evaluation consultation procedure to be carried out by an independent expert panel, based on the clinical evaluation assessment report of the same notified body.
In addition to the CE Marking, medical devices placed on the Italian market must be notified to the MoH and registered in an institutional register of medical devices.
4. What are the approximate fees for each authorization?
Both the EMA and the AIFA charge fees for applications for marketing authorization. Those due to the AIFA are established by the Decree of the Ministry of Health dated December 6, 2016. Said Decree also provides that, with respect to applications filed through the decentralized procedure where Italy acts as Reference Member State, fees are increased by 20%, whereas applications filed through the mutual recognition procedure where Italy acts as Reference Member State benefit from a 80% reduction. Fees are adjusted every year for inflation. Fee reductions and incentives are available for micro, small and medium-sized enterprises, designated orphan drugs, multiple applications on usage patent grounds and other classes of application.
5. For how long are marketing authorizations/registrations valid? How are marketing authorizations/registrations renewed?
Both national and centralized marketing authorizations for medicinal products are valid for five years. They can be renewed after 5 years from the first authorization on the basis of a re-evaluation of the risk-benefit ratio carried out by the AIFA or the EMA. Before the expiration of the validity of the MA, the relevant holder must submit a renewal application enclosing an updated version of the drug dossier covering the quality, safety and efficacy profiles. After renewal, the authorization is valid indefinitely, unless otherwise specified by the competent authority, which may decide to proceed with an additional five-year renewal.
6. How does the authorization process differ between brand-name products and generic products? Are there differences for local manufacturers versus foreign-owned manufacturers?
Under Italian law, generic drugs benefit from a simplified authorization procedure. Pursuant to the Pharma Code, a generic drug is a medicinal product which has the same qualitative and quantitative composition in active principles and the same pharmaceutical form of the reference medicinal product, and whose bioequivalence with the latter has been demonstrated by appropriate bioavailability tests. In other words, generic drugs are those medicinal products that, on the basis of predetermined, objective and measurable scientific and methodological criteria, can be regarded as equivalent, in terms of quality, safety and efficacy, to reference originator drugs developed and authorized following clinical studies which confirmed the relevant prerogatives.
More in particular, art. 10 of the Pharma Code establishes the principle that an applicant for a marketing authorization shall not be required to provide the results of pre-clinical tests and of clinical trials if he can demonstrate that the concerned product is a generic of a reference medicinal product which is, or has been, authorized in Italy of in the EU for at least 8 years. A generic drug so authorized shall not, however, be placed on the market until 10 years have elapsed from the initial authorization of the reference product (so-called “market exclusivity”); a clear reference to such a prohibition is included in the provision granting the relevant marketing authorization. Said 10-year period can be extended to a maximum of 11 years if, during the first 8 years of those ten years, the marketing authorization holder (“MAH”) obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies.
In this respect, it is noteworthy to mention that, under Italian law, the applicant for a marketing authorization for a generic drug may carry out tests necessary for submitting the file before the end of the exclusivity period of the originator without this being regarded as an infringement of the rules on the protection of industrial and commercial property. Notwithstanding the foregoing, art. 11 of Law Decree No. 158/2012, provides that generic drugs shall not be reimbursed by the NHS before the expiration of the patent/supplementary protection of the originator.
There is no difference for local manufacturers versus foreign-owned manufacturers.
7. How are combination products (drug + drug, drug + biologic, drug + device, biologic + device, drug + biologic + device) regulated?
Art. 1(8) of the MDR provides that any device which, when placed on the market or put into service, incorporates, as an integral part, a substance which, if used separately, would be considered to be a medicinal product, including a medicinal product derived from human blood or human plasma, and that has an action ancillary to that of the device, shall be assessed and authorized in accordance with the MDR. However, if the action of that substance is principal and not ancillary to that of the device, the integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No. 726/2004 on medicinal products for human use, as applicable. In that case, the relevant general safety and performance requirements set out in Annex I to the MDR shall apply as far as the safety and performance of the device part are concerned.
Under Article (9) of the MDR, any device which is intended to administer a medicinal product shall be governed by the MDR, without prejudice to the provisions of Directive 2001/83/EC and of Regulation (EC) No. 726/2004 with regard to the medicinal product. However, if the device intended to administer a medicinal product and the medicinal product are placed on the market in such a way that they form a single integral product which is intended exclusively for use in the given combination and which is not reusable, that single integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No. 726/2004, as applicable. In that case, the relevant general safety and performance requirements set out in Annex I to the MDR shall apply as far as the safety and performance of the device part of the single integral product are concerned.
Lastly, Article 1(10) of the MDR establishes that any device which, when placed on the market or put into service, incorporates, as an integral part, non-viable tissues or cells of human origin or their derivatives that have an action ancillary to that of the device shall be assessed and authorized in accordance with the MDR. In that case, the provisions for donation, procurement and testing laid down in Directive 2004/23/EC on human tissues and cells shall apply. However, if the action of those tissues or cells or their derivatives is principal and not ancillary to that of the device and the product is not governed by Regulation (EC) No. 1394/2007 on advanced therapy medicinal products, the product shall be governed by Directive 2004/23/EC. In that case, the relevant general safety and performance requirements set out in Annex I to the MDR shall apply as far as the safety and performance of the device part are concerned.
8. How is compliance with regulation monitored and evaluated? Is the regulatory regime comparable with the U.S. Food and Drug Administration or the European Medicines Agency expectations and requirements?
a. Medicinal products
In Italy, the AIFA is the authority responsible for monitoring compliance with the rules governing the development, manufacturing, labelling, marketing, distribution, import and advertising of medicinal products and, more in general, for ensuring that drugs placed on the Italian market are safe and do not pose risks to human health, also by preventing falsified medicinal products from entering the legal supply chain.
To this end, the AIFA may inspect plants and premises where medicinal products are manufactured, imported and stored, take samples of products and subject them to compliance checks, review and take copies of all relevant documents and require economic operators to implement corrective measures. In performing the above-mentioned functions, the AIFA can be supported by the MoH, the National Institute of Health, the Ministry of the Economic Development, the Italian Customs Agency as well as the Anti-adulteration and Health Units of the Carabinieri Corp.
The AIFA has the power to suspend, revoke or modify authorizations, order the total or partial closure of manufacturing plants, prohibit the sale or the use of drugs, withdraw products from the market and to confiscate medicinal products.
b. Medical devices
The MoH is the designated national competent authority and market surveillance authority for medical devices and in vitro diagnostic medical devices. Under applicable law, the MoH is responsible for enforcing the medical device legislation and for performing appropriate checks on the conformity characteristics and performance of devices including, where appropriate, a review of documentation and physical or laboratory checks on the basis of adequate samples. In this context, the MoH may require manufacturers to provide justifications as well as to implement corrective measures.
The MoH has the power to confiscate, destroy or otherwise render inoperable devices that present an unacceptable risk where it deems it necessary to do so in the interests of the protection of public health.
9. What is the potential range of penalties for noncompliance?
Violations of the legislation on medicinal products and medical devices can be punished with criminal and administrative sanctions. Depending on the type of breach, and unless the relevant fact constitutes a more serious offence, criminal sanctions may result in the imprisonment up to three years whereas administrative sanctions may result in the application of pecuniary fines up to EUR 300,000.
10. Is there a national healthcare system? If so, how is it administered and funded?
Yes. The Italian National Health System provides citizens with a uniform level of healthcare services throughout the country. It encompasses both hospital care, assistance by affiliated doctors, and the reimbursement of pharmaceutical products that are essential for health protection.
The Italian NHS is based on the following fundamental principles:
• public responsibility of health protection;
• universality and equity of access to health services;
• public funding through general taxation;
• portability of rights throughout the country and reciprocity of assistance with other Regions.
The NHS is administered by public bodies at different institutional levels, each of which contribute to the achievement of the objectives of protecting the health of citizens, including:
• the MoH, which is the central body;
• the National Institute of Health; and
• local health authorities.
Proceeds from national and regional taxes fund the NHS.
11. How does the government (or public) healthcare system function with private sector healthcare?
In Italy, the reference legislative framework for the provisions of medical services is Legislative Decree No. 502/1992 on “Reorganization of the health legislation”, as subsequently amended and supplemented (“Decree No. 502/1992”). Pursuant to Decree No. 502/1992, that applies both to public and private healthcare structures, the provision of medical services is subject to a prior authorization granted by the competent Regional health authority after having ascertained that the relevant healthcare structure meets the minimum, structural, technological and organization requirements laid down by Italian laws.
Private healthcare structures wishing to provide medical services on behalf of the NHS and to obtain the reimbursement by the same NHS of the services provided must be accredited with the Region in whose territory the healthcare structure is located and enter into a specific agreement establishing, amongst other things:
• the relevant health objectives and programs for integrating health services;
• the maximum volume of health services that the concerned private healthcare structure undertakes to ensure;
• the requirements of the health services to be performed, with specific regard to accessibility of care, clinical and organizational appropriateness, waiting times and continuity of care;
• the estimated overall fees for the agreed health services;
• the methods to ensure compliance with the remuneration threshold.
12. Are prices of drugs and devices regulated and, if so, how?
a. Medicinal products
Yes. Under Law No. 326/2003, the AIFA is the body in charge of negotiating the cost of medicines reimbursed by the NHS and, more generally, monitoring prices of medicinal products. As to pricing, medicines not reimbursed by the NHS are governed by Law Decree No. 87/2005, under which the MAH can freely determine the price although the AIFA must still ensure that: (i) price increases occur at least every two years (odd years); and (ii) increases do not exceed the inflation level.
With respect to reimbursement, under Law No. 326/2003, prices reimbursed by the NHS are established through a bargaining procedure between the AIFA and the concerned MAH in accordance with the modalities and criteria provided for by the Decree of the MoH dated August 2, 2019. The criteria for the evaluation of a medicine for reimbursement purposes are mainly the following: “added therapeutic value” that the medicinal product must ensure with respect to the main treatments to which it is compared; economic evaluation; availability, consumption and possible reimbursement of the concerned medicinal product in other countries; annual market shares that the MAH expects to acquire in the following 36 months; MAH’s production capacity and ability to manage unexpected events; assessment of the economic impact on the NHS; the patent status of the relevant medicinal product; etc.
The added therapeutic value of the medicine is assessed by the AIFA with the assistance of the Technical-Scientific Committee whereas the adequacy of the reimbursement price is evaluated with the support of the Price and Reimbursement Committee.
In successful negotiations, the parties enter into an agreement setting the relevant price and the conditions for the reimbursement. The price agreed by the AIFA and the MAH is the maximum selling price to the NHS. The agreement may also provide for an expenditure ceiling applicable to the concerned medicinal product. The price determined through the agreement is valid for a period of 24 months. Upon expiry, the agreement is automatically renewed for a further 24 months unless the parties agree to amend it within 60 days of the expiry date. The list of medicines reimbursed by the NHS is published on the Official Journal of the Italian Republic and is updated every six months. Conversely, if the outcome of the negotiation is negative, the medicinal product is put in Class C with no NHS reimbursement.
Simplified procedures for establishing the reimbursement price of generic and biosimilar drugs are available.
b. Medical devices
There are no specific provisions of law governing the pricing and reimbursement procedure for medical devices. The NHS normally purchases medical devices through public procurement procedures. In this respect, it is noteworthy to mention that the National Anti-Corruption Authority (“ANAC”), which is the Italian authority responsible for monitoring the award and execution of public contracts, has been entrusted with the task of identifying the “reference prices” for certain types of medicinal devices. References prices represent the maximum prices for the sale of the relevant medical devices to the NHS. Public supply contracts providing for higher prices are null and void.
13. How are drugs and devices used by patients paid for? What roles do public and private payers play?
a. Medicinal products
For reimbursement purposes, drugs are classified in the following three categories:
• “Class A” medicines, which include drugs for essential and chronic diseases that are entirely reimbursed by the NHS;
• “Class H” medicines, which include essential drugs fully reimbursed by the NHS for hospital use only; and
• “Class C” medicines, which include all other medicinal products that are not reimbursed by the NHS and for which citizens have to pay; and
• “Class Cnn” medicines, which include medicines not yet assessed for NHS reimbursement purposes.
Only physicians and healthcare structures belonging to or accredited with the NHS can prescribe and supply drugs reimbursed by the NHS.
b. Medical devices
Medical devices are paid directly by the NHS, through public tender procedures, or by private owners of hospitals.
14. Who dispenses drugs and devices to patients and how are those dispensers compensated?
Only authorized HCPs are allowed to dispense drugs and devices. They are compensated under applicable Collective Bargaining Agreements.
15. What are the professional and legal responsibilities of those who dispense drugs and devices? What role do they play in providing patient care, information, and safety?
HCPs must provide adequate patient care and information to patients and make sure that products are safely administered- Professional and legal responsibilities are those identified by their Professional Boards and by Italian criminal and civil law (i.e. medical malpractice legislation).
Also from this Legal Handbook
9. EU Ruling likely to set an increase in off-label use of medicinal products.
Managing partner of the law firm Herbert Smith Freehills in Italy, Laura Orlando is one of Italy’s leading life sciences specialists, with a focus on IP and regulatory matters. Together with Sara Balice, one of the firm’s senior associates they examine the Court of Justice of the European Union’s (CJEU) ruling on the Reimbursement of off-label medicinal products.
It is the responsibility of EU Member States to control off-label use of medicinal products.
Under EU Legislation (Directive 2001/83/EC, the “Directive”), a medicinal product may only be placed on the market of a Member State if a marketing authorisation has been issued by the competent authorities. The terms under which a medicinal product can be used safely and efficaciously are described in the product information, which is an integral part of the marketing authorisation process.
Off-label use refers to any intentional use of an authorised product not covered by the terms of its marketing authorisation.
A study report on the off-label use of medicinal products in the EU published by the European Commission in 2017 revealed that various drivers provoke off-label use of medicinal products. These drivers relate to the marketing authorisation process, post-marketing authorisation events (e.g. disruption in manufacturing, withdrawal from the market), pricing and reimbursement, aspects connected with the work of healthcare professionals, and patient-related factors, as shown in the figure below.
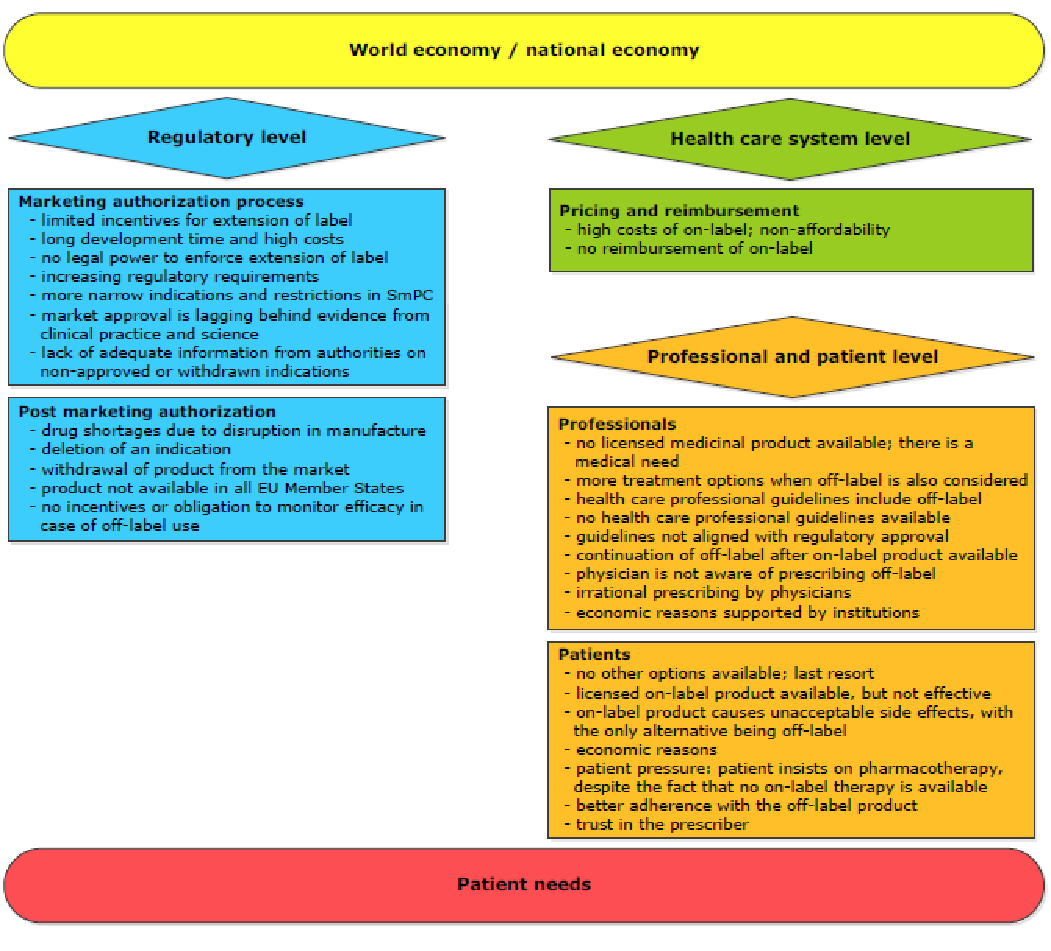
It is the responsibility of EU Member States to control off-label use of medicinal products. EU rules do not prohibit off-label prescription of medicines and their repackaging for such use but do require that they comply with the conditions laid down in those rules.
Italian legislation
Under Italian law, further to an amendment introduced in 2014 (Law 79/2014), off-label medicinal product can be reimbursed by the Italian national healthcare insurance system (NHS) not only if no other therapeutic alternative is available but also in order to reduce expenditure, notwithstanding the existence and market availability of a valid therapeutic alternative.
Avastin case
The above-mentioned Italian law has been the subject of a recent decision of the CJEU in Novartis Farma v Italian Medicines Agency (AIFA), Roche Italia et al. case (C-29/17). The Court answered, in essence, that such law is not contrary to EU law on medicinal products.
The case involved two medicinal products – Avastin and Lucentis – marketed, respectively, by Roche and Novartis. Avastin is authorised for cancer treatment and Lucentis for eye diseases. Avastin was also frequently used off-label to treat eye diseases and was included in the Italian list of authorised, reimbursable off-label products – known as “List 648”. When Lucentis was authorised for the treatment of eye diseases in 2012, Avastin was excluded from List 648.
In 2014, the Italian competition authority fined both Roche and Novartis for having created an artificial differentiation between Avastin and Lucentis when they were equivalent for the treatment of eye diseases. After the competition case, AIFA reinserted Avastin as reimbursable after repackaging by the pharmacies for its off-label treatment of eye diseases, because Avastin had to be extracted from its original vial and divided into single-use syringes for intravitreal injection. Italian Law 79/2014 was then introduced, which essentially granted AIFA the power to authorise off-label use of medicinal products (under certain conditions) if warranted by public interest (including for financial reasons) even where a valid therapeutic alternative was available.
Novartis challenged AIFA’s decision to insert Avastin in List 648 but the decision was upheld by the Regional Administrative Court for Lazio. Novartis lodged an appeal with the Italian Council of State, which referred the case to the CJEU. The Italian Council of State essentially asked for clarification on whether:
- a national law, which encourages off-label use of medicinal products – regardless of specific patient needs and even when an authorised medicinal product for the same treatment already exists – by including medicinal products in the national list of NHS-reimbursed medicinal products, is in contrast with the Directive;
- once Avastin has been repackaged for off-label use according to national law, it falls within the scope of the Directive;
- a national law, which grants AIFA the power to monitor drugs like Avastin for its off-label use is in contrast with Regulation (EC) 726/2004.
CJEU Decision
The CJEU decision ruled that:
- The repackaging of Avastin in pharmacies for the purpose of its off-label use does not significantly change its composition, form or other fundamental characteristics. Therefore, repackaged Avastin still falls within the scope of the Directive.
- The Directive does not impact the national law that allows Avastin to be repackaged for its off-label treatment of ophthalmological indications, provided that the process of repackaging is prescribed by a doctor via an individual prescription and carried out by pharmacists.
- Regulation (EC) 726/2004 does not conflict with national law, under which AIFA has the power to monitor off-label products to safeguard patient safety, provided that its implementation reinforces the pharmacovigilance system introduced by Regulation No 726/2004.
This decision confirms that the Directive prohibits neither off-label use nor the repackaging of medical products for off-label use provided that it complies with the Directive. However, the CJEU did not expressly address the question whether off-label use of a medicinal product is permitted under the Directive for the sole purpose of reducing the NHS spend when medicinal products authorised for that specific therapeutic indication are already available on the market. The position of the Advocate General was clearer as he opined that (i) off-label use should be permitted insofar as it is necessary for therapeutic reasons (even when an authorised medicinal product for the same treatment already exists), and (ii) a national law that allows physicians to prescribe off-label products for the sole purpose of cost-saving would be contrary to the Directive.
It is likely that in the circumstances we will see an increase in off-label use.