The Pharma Legal Handbook: Spain





 To navigate its highly promising, but complex and regionalised environment, expert insight is crucial. To this end, The Pharma Legal Handbook: Spain is a comprehensive legal guide that answers essential questions about the legal and regulatory environment for pharmaceutical companies in the country. It is a must-have for any company operating in the country or looking to enter the market.
To navigate its highly promising, but complex and regionalised environment, expert insight is crucial. To this end, The Pharma Legal Handbook: Spain is a comprehensive legal guide that answers essential questions about the legal and regulatory environment for pharmaceutical companies in the country. It is a must-have for any company operating in the country or looking to enter the market.
Prepared in association with Faus & Moliner Abogados, a leading Spanish law firm.
August 2022
1. Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs: Spain
Cannabinoid drugs, medicinal cannabis and opioid drugs in Spain – a legal guide. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
Cannabinoid Drugs
For the purposes of this Chapter 8: Cannabinoid Drugs, Cannabinoid Drugs shall mean any industrially produced medicinal product which contains cannabinoids.
1. Are Cannabinoid Drugs authorized in your country?
Cannabinoid Drugs may be authorized in Spain on the basis of scientific criteria on its quality, safety and efficacy. The foregoing is provided that applicable regulations on narcotics, such as Law 17/1967 on narcotics, and Royal Decree 2829/1977 on narcotics and its production, distribution, prescription and dispensation are observed.
2. What are the regulatory authorities with jurisdiction over Cannabinoid Drugs?
The main Spanish authorities with jurisdiction over Cannabinoid Drugs are the Spanish Ministry of Health and the AEMPS.
3. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Cannabinoid Drugs?
No. The general regulatory framework applicable for the authorization, pricing and reimbursement of medicinal products apply to Cannabinoid Drugs.
There are, however, specific regulations that may be applicable to other activities related to Cannabinoid derivatives whenever they are considered narcotics, such as manufacturing, distributing, prescribing, dispensing etc. (see, among others, Law 17/1967, Royal Decree 2829/1977 and Royal Decree 1675/2012 on prescribing and dispensing narcotics for both human and veterinary use).
4. Which are the cannabinoid drugs that have received market approval to date?
To date, only Sativex 2,7 mg / 2,5 mg Oromucosal spray solution (“Sativex®”) has received market approval in Spain. Sativex® is a Cannabinoid Drug which contains, per each single 100 microlitre spray, 2.7 mg delta-9-tetrahydrocannabinol (THC) and 2.5 mg cannabidiol (CBD) from Cannabis sativa L. The marketing authorization holder of Sativex is GW Pharma Ltd.
Sativex® has been classified as a hospital diagnosis drug, which means that it shall only be used for the treatment of diseases diagnosed either in hospitals or centres which have appropriate diagnosis means, or by specialized physicians (see art. 24.3.b of Royal Decree 1345/2007 on the procedure for authorization of industrially produced medicinal products for human use).
Likewise, according to section 4.1 of the technical sheet of Sativex®, Sativex® must only be used “to improve symptoms of adult patients with moderated or serious spasticity due to multiple sclerosis” if “other anti-spasticity medicines have not worked” and “a significant clinical improvement has been observed regarding the symptoms of spasticity after an initial trial period of the treatment”.
5. Who can prescribe Cannabinoid Drugs?
Physicians, odontologists and veterinaries, each of them in the field of its competence, can prescribe narcotic drugs.
6. Is there a list of doctors authorized to prescribe Cannabinoid Drugs?
There is not a list of doctors authorized to prescribe Cannabinoid Drugs. Doctors, provided that they comply with the applicable requirements and procedures (which include the need to use the appropriate prescription forms), can prescribe Cannabinoid Drugs without need to be previously registered in any official list. It is to be noted that doctors holding prescription forms for narcotic drugs may be controlled (and, therefore, somehow listed) but these are lists relating to the prescription forms (holders of such forms are listed) and not related to the doctors themselves.
7. What approvals or notifications are required to prescribe Cannabinoid Drugs?
Provided that applicable regulations are complied with (including the use of the appropriate prescription forms), doctors themselves do not have to ask for any specific approval or make any specific notification each time they prescribe Cannabinoid Drugs.
8. Which organizations are authorized to sell/distribute Cannabinoid Drugs available?
Cannabinoid Drugs (provided that their dispensation to the public is not specifically restricted in their marketing authorization) can be dispensed to the public by authorized pharmacy offices/services.
Cannabinoid Drugs can be distributed (that is, sold to either other authorized distribution entities or pharmacy offices/services) by any of the following entities provided that they comply with all applicable regulations on medicinal products and narcotics (such as Law 17/1967, Royal Decree 2829/1977 -and its complementary regulations- and Royal Decree 1675/2012):
- Pharmaceutical laboratories;
- Distribution entities duly authorized to distribute medicines for human use (see Royal Decree 782/2013 on distribution of medicinal products for human use); and/or
- Distribution entities duly authorized to distribute veterinary medicines (see Royal Decree 109/1995 on veterinary medicinal products).
9. Is there a list of retailers/distributors authorized to sell Cannabinoid Drugs?
- Retailers. The AEMPS, with the cooperation of the regional authorities, publishes lists including all pharmacy offices opened to the public in Spain. Such pharmacy offices can dispense Cannabinoid Drugs if they comply with applicable regulations (please refer to Question 8).
- Distributors. The AEMPS publishes lists including all entities authorized to distribute veterinary medicines and medicines for human use in Spain. All entities authorized to distribute Cannabinoid Drugs must be included in such lists.
10. Are there proposals for reform or significant change to the regulation of Cannabinoid Drugs?
We are not aware of any proposal to significantly reform the regulation of Cannabinoid Drugs in Spain. Spanish Law on Medicinal Products, which is the basic regulatory framework for medicinal products, is subject to a process of modification and currently in the phase of public consultation. However, we have not detected, within the objectives pursued by this process of amendment, any reference to the regulatory regime of Cannabinoid Drugs. Please refer to Question 24 for further information regarding Medicinal Cannabis.
11. When are they likely to come into force?
N/A.
Medicinal Cannabis
For the purposes of this Chapter 8: Medicinal Cannabis, Cannabis shall mean “the flowering or fruiting tops of the cannabis plant (excluding the seeds and leaves when not accompanied by the tops) from which the resin has not been extracted” (see art. 1 of the Single Convention on Narcotic Drugs of 1961); and Medicinal Cannabis shall mean Cannabis used/intended to be used for medicinal purposes. Cannabinoid Drugs (as defined in Chapter 8 above), products formulated by pharmacists containing cannabinoids which qualify as medicines under Spanish law (see art. 8.1.b and c of the Spanish Law on Medicinal Products) and cannabinoid products with very low concentrations of THC fall outside the concept of Medicinal Cannabis for the purposes of this Chapter.
12. Is Medicinal Cannabis authorized in the country?
The use of Cannabis for medicinal purposes is not specifically regulated in Spain; being a legal void in this field which generates significant legal uncertainty.
Notwithstanding the foregoing, we make the following comments:
- Cannabis qualifies as a narcotic in accordance with art. 2.1 of Law 17/1967 and List I of the Single Convention on Narcotic Drugs of 1961. Likewise, Cannabis is considered as a “prohibited product” in accordance with art. 2.2 of Law 17/1967 and List IV of the Single Convention on Narcotic Drugs of 1961.
- Art. 22 of Law 17/1967 (which states that the use of narcotic drugs for therapeutic uses may be allowed under certain circumstances), art. 51.3 of the Spanish Law on Medicinal Products (which states that plants traditionally used as medicines and offered without reference to therapeutic, diagnosis or preventive properties may be sold under certain circumstances)and other regulations have been used by certain authors to try to defend Medicinal Cannabis in Spain. Such interpretations, however, shall be read with extreme caution (and be considered as mere opinions) as long as in Spain there is not a clear regulatory framework regarding Medicinal Cannabis and, therefore, no clear and solid conclusions can be reached in this regard.
- From a criminal point of view, self-consumption of Medicinal Cannabis (provided that the corresponding requisites established by case law are complied with) is not criminally punishable. Other activities connected somehow with Medicinal Cannabis shall be carefully analyzed as long as they may trigger legal risks, including criminal ones (see art. 348 of the Spanish Criminal Code which literally states that “those who execute acts of cultivation, processing or trafficking, or otherwise promote, favour or facilitate the illegal consumption of toxic drugs, narcotics or psychotropic substances, or possess them for those purposes, shall be punished…”).
- Rules such as Law 17/1967, Royal Decree 2829/1977 (and its complementary regulations), Royal Decree 1675/2012 and criminal regulations should be considered when reviewing rules applicable to Medicinal Cannabis as long as certain activities connected somehow with Medicinal Cannabis may fall within their scope.
- With regard to the CBD as a cannabinoid derived from cannabis, the question about its legality was put in front of the Court of Justice of the EU (CJEU). On 19 November 2020, the CJEU decided in the “Kannavape case” that the rules on the free movement of goods within the European Union (articles 34 and 36 of the Treaty on the Functioning of the European Union, TFEU) “must be interpreted as precluding national legislation which prohibits the marketing of CBD lawfully produced in another Member State when it is extracted from the Cannabis sativa plant in its entirety and not solely from its fibre and seeds”. According to the CJEU, CBD does not qualify as a narcotic drug in that matter, since it appears that the CBD in that matter (used for e-cigarettes) does not have any psychotropic or harmful effect on human health. This resolution confirming that CBD is not a narcotic is fully applicable in Spain.
It is to be noted that Medicinal Cannabis is being object of discussion for regulation within the Spanish Congress of Deputies. Please refer to Question 24.
13. What are the regulatory authorities with jurisdiction over Medicinal Cannabis?
The main Spanish authorities with jurisdiction over Medicinal Cannabis are the Spanish Ministry of Health and the AEMPS.
14. What is the regulatory framework for the authorization, pricing, and reimbursement of Medicinal Cannabis?
There is no special regime for Medicinal Cannabis and consequently, for its authorization, pricing and reimbursement. With regard to Cannabis Drugs, please refer to Question 12.
15. How is the production and import of Medicinal Cannabis regulated and by which agencies/authorities?
Production/import of Cannabis is mainly regulated in Law 17/1967 and Royal Decree 2829/1977. General foreign trade regulations and international conventions, when applicable, shall also be observed.
In addition to the foregoing, it is also worth mentioning a ministerial order dated on 7 May 1963 on cultivation of medicinal plants related to narcotics and two informative notes published in the official website of the AEMPS in December 2018 which regulate the matter of the cultivation of Cannabis for medicinal and scientific purposes. According to these notes, cultivation of Cannabis for these purposes requires a previous authorization from the AEMPS. The proceeding for this authorization to be granted has not been laid down in any specific law other than the informative notes and has also been widely criticised for lacking transparency and clarity.
The Spanish Ministry of Health and the AEMPS have authority regarding the production/import of Cannabis.
16. What approval or notifications are necessary to produce or import Medicinal Cannabis?
The production/import of Cannabis is only permitted for scientific and/or medicinal purposes and it is subject to an authorization to be granted by Spanish regulatory authorities according to the regulations and the informative notes mentioned in Question 15.
17. What is the regulatory framework for the marketing and distribution of Medicinal Cannabis?
There is no regulation specifically regulating the promotion of Medicinal Cannabis. However we make the following comments:
- If a product (such as Cannabis) is presented for treating or preventing diseases, it may be considered as a medicinal product according to Spanish regulations (see art. 2.a of Spanish Law on Medicinal Products which refers to the definition of medicinal product by presentation);
- In Spain, medicinal products cannot be promoted if they have not received a valid marketing authorization (see art. 2.1 Royal Decree 1416/1994 on marketing of medicinal products for human use).
- Provided that Cannabis, to date, has not received a marketing authorization, the promotion of Medicinal Cannabis is not acceptable.
- It is also worth mentioning the fact that plants that have been traditionally considered as medicinal plants (someone could try to argue that this is the case of cannabis) have a specific regime in Spain (see art. 51.3 of Spanish Law on Medicinal Products and related regulations). Without analyzing such regime (or analyzing if cannabis can be authorized through the same), we note that such “medicinal plants”, even if they are finally authorized, cannot be “offered to the public with reference to its therapeutic properties”.
Regarding the distribution of Medicinal Cannabis, please refer to Question 12.
18. How can patients obtain Medicinal Cannabis?
Please refer to Question 12.
19. Who can prescribe Medicinal Cannabis?
Please refer to Question 12.
20. Is there a list of doctors authorized to prescribe Medicinal Cannabis?
Please refer to Question 12.
21. What approvals or notifications are required to prescribe Medicinal Cannabis?
Please refer to Question 12.
22. Where is Medicinal Cannabis available?
Please refer to Question 12.
23. Is there a list of retailers authorized to sell Medicinal Cannabis?
Please refer to Question 12.
24. Are there proposals for reform or significant change to the regulation of Medicinal Cannabis?
The Spanish Parliament (Congreso de los Diputados) has recently created a commission devoted to the analysis of the available scientific evidence in connection with the use of Medicinal Cannabis as well as the results of experiences of use. This commission issued, in June 2022, a report confirming that Medicinal Cannabis could have positive therapeutic effects for certain pathologies. This report also proposed the legal qualification that Medicinal Cannabis products may adopt (i.e. medicinal product, magistral formulas, etc.) or how this resulting products should be commercialized and distributed in Spain. We are not aware of any specific regulation being implemented yet. However, as per the information informally provided by the representatives of this commission, the issuance of this favourable report is the first step for the future regulation of Medicinal Cannabis, which should be expected between 2023-2024.
Opioid Drugs
For the purposes of this Chapter 8: Opioid Drugs, Opioid Drugs shall mean any industrially produced medicinal product which contains opioids.
25. Are Opioid Drugs authorized in your country?
Opioid Drugs may be authorized in Spain on the basis of scientific criteria on its quality, safety and efficacy. The foregoing is provided that applicable regulations on narcotics, such as Law 17/1967 on narcotics, and Royal Decree 2829/1977 on narcotics and its production, distribution, prescription and dispensation, are observed.
26. What are the regulatory authorities with jurisdiction over Opioid Drugs?
The main Spanish authorities with jurisdiction over Opioid Drugs are the Spanish Ministry of Health and the AEMPS.
27. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Opioid Drugs?
No. The general regulatory framework applicable for the authorization, pricing and reimbursement of medicinal products apply to Opioid Drugs.
There are, however, specific regulations that may be applicable to other activities related to Opioid Drugs such as manufacturing, distributing, prescribing, dispensing etc. (see, among others, Law 17/1967, Royal Decree 2829/1977 and Royal Decree 1675/2012 on prescribing and dispensing narcotics for both human and veterinary use).
28. Which are the Opioid drugs that have received market approval to date?
There are many medicinal products containing opioids that have received market approval to date in Spain.
An updated and complete list of all medicinal products authorized in Spain classified by active ingredient (as well as by trademark, registration number, etc) can be found in CIMA, and online data base held by the AEMPS and available in the official website of the AEMPS. All medicinal products containing opioids that have received market approval in Spain are included in such list.
29. Who can prescribe Opioid Drugs?
Doctors, odontologists and veterinaries, each of them in the field of its competence, can prescribe Opioid Drugs.
30. Is there a list of doctors authorized to prescribe Opioid Drugs?
There is not a list of doctors authorized to prescribe Opioid Drugs. Doctors, provided that they comply with the applicable requirements and procedures (which include the need to use the appropriate prescription forms), can prescribe Opioid Drugs without need to be previously registered in any official list. It is to be noted that doctors holding prescription forms for narcotic drugs may be controlled (and therefore somehow listed), but these are lists relating to the prescription forms (holders of such forms are listed) and not related to the doctors themselves.
31. What approvals or notifications are required to prescribe Opioid Drugs?
Provided that applicable regulations are complied with (including the use of the appropriate prescription forms), doctors themselves do not have to ask for any specific approval or make any specific notification each time they prescribe Opioid Drugs.
Treatments with Opioid Drugs for patients addicted to Opioid Drugs have their specific regulations (such as Royal Decree 75/1990 on treatments with opiates for people addicted to opioids, and the circular of the AEMPS 6/2003 regarding the conditions to distribute narcotics for the treatment of people addicted to opioids.
32. Which organizations are authorized to sell/distribute Opioid Drugs available?
Opioid Drugs can be dispensed to the public by authorized pharmacy offices/services.
Opioid Drugs can be distributed (that is, sold to either other authorized distribution entities or pharmacy offices/services) by any of the following entities provided that they comply with all applicable regulations on medicinal products and narcotics (such as Law 17/1967, Royal Decree 2829/1977 -and its complementary regulations- and Royal Decree 1675/2012):
- Pharmaceutical laboratories;
- Distribution entities duly authorized to distribute medicines for human use (see Royal Decree 782/2013); and/or
- Distribution entities duly authorized to distribute veterinary medicines (see Royal Decree 109/1995).
33. Is there a list of retailers/distributors authorized to sell Opioid Drugs?
Retailers. The AEMPS, with the cooperation of the regional authorities, publishes lists including all pharmacy offices opened to the public in Spain. Such pharmacy offices can dispense Opioid Drugs if they comply with applicable regulations (please refer to Question 32).
Distributors. The AEMPS publishes lists including all entities authorized to distribute veterinary medicines and medicines for human use in Spain. All entities authorized to distribute Opioid Drugs must be included in such lists.
34. Are there proposals for reform or significant change to the regulation of Opioid Drugs?
We are not aware of any proposal to significantly reform the regulation of Opioid Drugs in Spain.
35. When are they likely to come into force
N/A.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
2. Regulatory Reforms : Spain
Want to know more about regulatory reforms in Spain? Read on! Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
1. Are there proposals for reform or significant change to the healthcare system?
In July 2022, the Ministry of Health opened a public consultation on the first draft of the law that will amend the current Spanish Law on Medicinal Products. The document published by the Ministry of Health shows that the reform that is being considered will have three principal axes:
a) Public financing of medicines
The document of the Ministry of Health refers to adopting new measures to rationalize pharmaceutical expenditure and promote rational use of public funds. In this regard, it is proposed to modify the Reference Price System by introducing elements that increase competition and value the contributions that represent an incremental benefit in the use of medicines. The document contemplates modifying the system of co-payment of medicines with the purpose of protecting the persons that are more in need. The document does not refer to whether the co-payment system may also be used as an instrument that may help modulating the demand of certain products. The document also announces measures of additional pressure to the industry by stating that quarterly contributions may also apply to medicines dispensed in healthcare centres.
b) The experience of the pandemic and the impact of new technologies
The pandemic has created important challenges related to the availability of medicinal products and medical devices. In this sense, the Ministry of Health aims to consolidate the non-presential dispensing of medicines for hospital dispensing and telepharmacy in the National Health System.
c) Implementation of the European Union Law
The text published by the Ministry of Health proposes to make the necessary amendments to incorporate the amendments and definitions of the Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro medical devices into Spanish law.
d) Clarify competences regarding the control of advertising of medicinal products and medical devices.
The aim of this reform is to undertake a comprehensive regulation of the advertising of medicinal products for human use and medical devices for both the general public and healthcare professionals. It also aims to better define the competences of the State and the Autonomous Communities in the different areas of action in the field of advertising. Finally, the aim is to adapt the regulations to technological advances, in particular to the predominance of digital and audiovisual media.
e) Modify and update the sanctioning procedure and the infringements provided for in Royal Legislative Decree 1/2015.
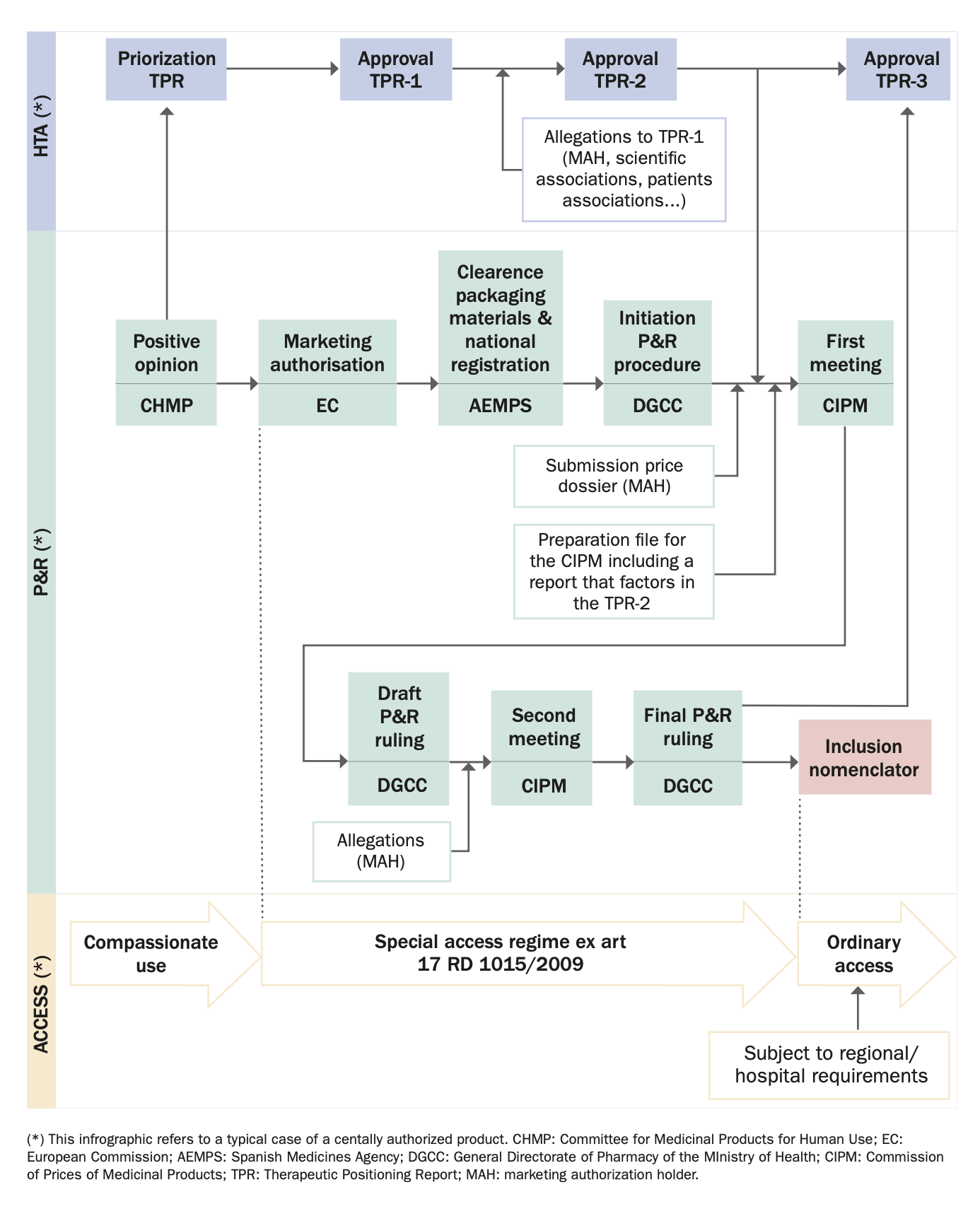
On the other hand, it is also proposed to reform Royal Decree 1015/2009. The aim of this reform is to better delimit the different existing cases of access to medicines in special situations and the different categories included in each of them, as well as the healthcare setting in which they can be used. It also aims to improve the procedure for accessing them. On the other hand, the aim is to introduce elements that guarantee that the use of medicinal products in special situations does not become routine, establishing measures that encourage the marketing and use of medicinal products through the channels established in ordinary legislation without the exceptional becoming habitual.
A draft of this new Royal Decree has not yet been published.
2. When are they likely to come into force?
The process for approval of the abovementioned proposals is still ongoing. Considering the current political situation in Spain (near elections at a national and local level), it is difficult to predict when such proposals will be approved and how the final texts of the same will be.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
3. Patents and Trademarks: Spain
The legal framework for patents and trademarks in Spain. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
1. What are the basic requirements to obtain patent and trademark protection?
Pursuant to Law 24/2015 on patents, the basic legal requirements to obtain a patent are: (i) to have a new invention in any field of technology; (ii) that such invention involves an inventive step; and (iii) that such invention is susceptible of industrial application.
Pursuant to Law 17/2001 on trademarks, the basic legal requirement to obtain a trademark is to have a sign subject of graphic representation which has a distinctive character and which is not identical with or similar to earlier trademarks, in order to avoid likelihood of confusion on the part of the public.
2. What agencies or bodies regulate patents and trademarks?
The Spanish Patent and Trademark Office (www.oepm.es).
3. What products, substances, and processes can be protected by patents or trademarks and what types cannot be protected?
The products, substances, and processes that can be protected by patents are those which can be considered as ‘inventions’, meeting the basic legal requirements referred to in Chapter 6, Question 1, which are not subject to any of the following exceptions to patentability:
- inventions the commercial exploitation of which would be contrary to public order or morality principles, including but not limited to processes for cloning human beings, processes for modifying the germ line genetic identity of human beings, uses of human embryos for industrial or commercial purposes, or processes for modifying the genetic identity of animals which are likely to cause them suffering without any substantial medical benefit to man or animal, and also animals resulting from such processes;
- plant or animal varieties; however, it is possible to patent plant or animal varieties if the technical feasibility of the invention is not confined to a particular plant or animal variety;
- essentially biological processes for the production of plants or animals, meaning those processes which consist entirely of natural phenomena such as crossing or selection; however, it is possible to protect by patent inventions consisting in microbiological or other technical process, or a product obtained by means of such processes;
- methods for treatment of the human or animal body by surgery or therapy and diagnostic methods practised on the human or animal body; however, it is possible to patent products, in particular, substances or compositions, for use in any of these methods;
- the human body, at the various stages of its formation and development, and the simple discovery of one of its elements, including the sequence or -partial sequence of a gene; however, it is possible to patent an element isolated from the human body or otherwise produced by means of a technical pro-cess, including the sequence or partial sequence of a gene, even if the structure of such element is identical to that of a natural element; or
- a mere DNA sequence, without indicating any biological function.
The law does not define the term ‘invention’ but it states what cannot be considered as such. To such effect, the following cannot be regarded as inventions:
- discoveries, scientific theories and mathematical methods;
- literary, artistic or scientific works or any other aesthetic creations;
- schemes, rules and methods for performing mental acts, playing games or doing business, and programs for computers; or
- ways to present information.
On the other hand, trademark protection is possible for any signs meeting the basic legal requirements referred to in Question 1, provided that such signs do not fall within any of the following prohibitions:
- those consisting exclusively of signs or indications which may serve, in trade, to designate the kind, quality, quantity, intended purpose, value, geographical origin, or the time of production of the goods or of rendering of the service, or other characteristics of the goods or services;
- those consisting exclusively of signs or indications which have become customary in the current language or in the bona fide and established practices of the trade;
- signs consisting exclusively of: (i) the shape, or another characteristic, which results from the nature of the goods themselves; (ii) the shape, or another characteristic, of goods which is necessary to obtain a technical result; (iii) the shape, or another characteristic, which gives substantial value to the goods;
- signs which are contrary to public order or morality principles;
- signs which are deceiving the public, e.g. to the nature, quality or geographical origin of the goods or service;
- signs which have not been authorized by the competent authorities and are to be refused or invalidated;
- signs copying or imitating the armorial bearings, flag, insignias and other emblems of Spain, its Autonomous Regions, municipalities, etc.
4. How can patents and trademarks be revoked?
Pursuant to article 102 of the Law 24/2015, a patent can be invalidated based on any of the following grounds:
- if the subject matter of the patent is not patentable, (please refer to Questions 1 and 3);
- if the invention is not disclosed in a manner sufficiently clear and complete for it to be carried out by a person skilled in the art;
- if its subject-matter exceeds beyond the content of the application as filed, or, if the patent was granted on a divisional application or on a new application filed by a non-entitled person, beyond the content of the earlier application as filed;
- if the protection of the patent was extended after its grant; or
- if the holder of the patent is not its rightful owner as provided in the law.
Pursuant to the provisions contained in article 105 of Law 24/2015, the holder of a patent can request the Spanish Patent and Trademark Office to revoke the patent at any time during its validity. To such effect, an official form must be filled-out, and the corresponding fees paid. The authorities can deny such request in certain cases, for example, if the patent is subject to certain type of rights (such as rights in rem or call options) or licenses dully recorded in the Patent Registry.
On the other hand, a trademark can be revoked due to failure to renew it, withdrawal by the owner or failure to pay the maintenance fees. Also, according to article 54 of Law 17/2001, a trademark can be revoked pursuant to a request filed before the Spanish Patent and Trademark Office or a counterclaim in trademark infringement proceedings, based on any of the following grounds:
- if during a five-year period the trademark has not been put to genuine use in Spain for the products or services for which it has been registered;
- if, due to acts or inactivity of the proprietor, the trademark has become the common name in the trade for a product or service in respect of which it is registered; or
- if, due to the use of the trademark by the proprietor or with the proprietor’s consent in respect of the goods or services for which such trademark is registered, if can mislead the public, particularly as to the nature, quality or geographical origin of those goods or services.
5. Are foreign patents and trademarks recognized and under what circumstances?
Both patents and trademarks are territorial rights, meaning that they are only applicable and enforceable in the country in which they have been filed and granted. Therefore, in order to be recognized in Spain, patents and trademarks must be validated / registered before the Spanish Patent and Trademark Office. In this regard, pursuant to the European Patent Convention, a European patent validated in Spain has the same effect of and is subject to the same conditions as a Spanish national patent. Regarding Unitary Patents, Spain is one of the few EU member states who did not ratify the Unified Patent Court Agreement. Therefore, neither Unitary Patents nor Unitary Patent Court decisions will have effects in Spain.
As regards trademarks, it is possible to fill a request for an EU trademark before European Union Intellectual Property Office. The EU trademark has a unitary character and, thus, is effective throughout the entire territory of the European Union.
Also, according to the so-called Madrid System, which includes two international treaties: the Madrid Agreement and the Madrid Protocol, it is possible to fill an international trademark request. Such request can be filed before the Spanish Patent and Trademark Office, in case the applicant is Spanish or based in Spain. The protection granted will be applicable in countries belonging to the Madrid System (currently 119 countries, including Spain).
6. Are there any non-patent/trademark barriers to competition to protect medicines or devices?
Yes, there are non-patent/trademark barriers to competition when it comes to protecting medicinal products in Spain.
On the one hand, there is the data exclusivity period regulated under Spanish Law on Medicinal Products. The data exclusivity period is the one during which the applicant of a generic medicinal product cannot cross-reference the clinical data (i.e. results of pre-clinical tests and clinical trials) of the reference medicinal product. This period lasts 8 years from the approval of the first marketing authorization of the reference medicinal product by the authorities of any Member State (not necessarily Spain) or the European Union. Once this 8-year period has expired, the applicant of a marketing authorization for a generic medicinal product is not required to provide the results of pre-clinical tests and clinical trials.
On the other hand, there is another barrier to competition related to medicinal products known as the market exclusivity period. Once a generic medicinal product is authorized using the results of pre-clinical tests and clinical trials of the reference medicinal product, it cannot be launched until the expiry of the marketing exclusivity period of 10 years counted from the first marketing authorization of the reference medicinal product in the EU. The exclusivity period of 10 years may be extended for 1 additional year, i.e. up to 11 years maximum, if, during the first 8 of those 10 years, the holder of the marketing authorization of the reference medicinal product obtains an approval for new indication(s) having significant clinical benefit in comparison with existing therapies.
As regards orphan medicinal products, there is a market exclusivity period conferred by Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products, which is directly applicable in Spain. Pursuant to the provisions contained in article 8 of this Regulation, when there is a marketing authorization granted in Europe for an orphan drug, the authorities in Europe and in all Member States must refrain during a 10-year period from accepting another marketing authorization application or from granting another marketing authorization, when there is an existing marketing authorization for a similar medicinal product having the same therapeutic indication. Such 10-year market exclusivity period may be shortened to 6 years if, at the end of year 5, it is established that the product no longer meets the criteria laid down for granting the orphan designation in the first place, and it is proven that such product is sufficiently profitable not to justify maintenance of market exclusivity. However, the market exclusivity period for orphan drugs will not apply, and therefore another marketing authorization may be granted for a similar medicinal product having the same therapeutic indication if:
- the holder of the marketing authorization for the original orphan medicinal product has given his consent to the second applicant, or
- the holder of the marketing authorization for the original orphan medicinal product is unable to supply sufficient quantities of the medicinal product, or
- the second applicant can establish in the application that the second medicinal product, although similar to the orphan medicinal product already authorized, is safer, more effective or otherwise clinically superior.
7. Are there restrictions on the types of medicines or devices that can be granted patent and trademark protection?
Not specifically. Please refer to Question 3.
8. Must a patent or trademark license agreement with a foreign licensor be approved or accepted by any government or regulatory body?
No, Spanish government or regulatory bodies do not need to approve or accept patent/trademark license agreements.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
4. Product Liability: Spain
The low-down on the situation regarding product liability in Spain. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
1. What types of liability are recognized in your jurisdiction?
The Spanish regime for product liability is a strict liability regime. Such regime imposes strict liability to the “producer” of a defective product. The producer will be liable for personal injury or death or damage to property caused by the defective product, provided that such defective product was objectively intended for private use or consumption and was utilized mainly as such by the injured party. It is on the claimant to prove that (i) the product was defective, (ii) the damage occurred and (iii) there was a causal link between the defective product and the damage suffered.
This strict liability system does not preclude other liability systems providing the injured party with a greater protection neither affects any other right to compensate damages, including moral damages, that the injured party may have as a consequence of contractual liability, based on the lack of conformity of the goods or any other cause of non-performance or defective performance of the contract, or of any non-contractual liability that may apply.
2. How do these types of liabilities apply to the manufacturers of medicines and devices?
Under the Spanish product liability regime only the “producer” bears responsibility for the fault of the product.
It is considered as “producer”, depending on the case, any or all of the followings: (i) the manufacturer or the importer in the European Union of a finished product, raw material or component of the product; and (ii) the apparent producer of the product (i.e. any person presenting itself as the producer of the product by providing its name, trademark or other identifying features along with the product, whether on the container, wrapping or other any protective or presentational component).
In the event that the “producer” cannot be identified, the supplier of the product (i.e. the distributor or the “retail” supplier) shall be considered as such, unless it informs the injured party about the identity of the manufacturer or of the person who supplied the product to it, within a term of three months before it is requested to give such information. This same rule applies in the case of imported products in the European Union, if the product does not indicate the name of the importer, even if it indicates the name of the manufacturer.
The supplier of a defective product shall also be liable towards the injured party as if it was the producer if it supplied the product knowing that the defect existed.
Therefore, according to Spanish law not only the final manufacturer, or the importer in EU, of a medicinal product or a medical device will be subject the Spanish product liability regime. Such regime will also apply to the manufacturer, or importer in the EU, of raw material or component of said product as well as to the apparent producer of said medicinal product or medical device and to the supplier under certain circumstances.
3. Does potential liability extend to the manufacturer only or could claims extend to corporate executives, employees, and representatives?
Under the Spanish regime on liability for defective products, the responsibility for damages caused by a defective product is only borne by the “producer” [i.e. (i) the manufacturer or the importer who introduces the product into the European Union; (ii) the apparent producer and; (iii) the supplier only under certain circumstance]. Therefore, as the corporate executives, employees, and representatives are not the producer, they will not be responsible under this regime.
4. How can a liability claim be brought?
A liability claim shall be brought by filing a claim before the Courts, by every injured party. When the injured party is a group of consumers, then the claim can be brought by associations of consumers, a group of consumers or by the Public Prosecutor’s Office.
5. What defenses are available?
The producer shall not be liable if they can prove that the product is not defective because it provides the safety which could legitimately be expected from it, taking all circumstances into account, including the time when the product was put into circulation, the presentation of the product and the reasonable use of the product.
Additionally, the producer shall not be liable if it can prove that:
- it did not put the product into circulation;
- given the circumstances of the case, it may be presumed that the defect did not exist when the product was put into circulation;
- the product had not been manufactured for sale or for any other form of distribution with an economic purpose, nor that it was manufactured, imported, supplied or distributed within the context of a professional or entrepreneurial activity;
- the defect is due to the fact that the product was manufactured in accor-dance with existing mandatory rules; and
- the state of scientific and technical knowledge existing at the time the product was put into circulation did not allow for the discovery of the existence of the defect.
The producer of a part that is integrated into a finished product shall not be liable if it proves that the defect is attributable to the design of the product into which the part was integrated, or to the instructions provided by the manufacturer of the finished product.
Additionally, the doctrine points out that the apparent producer shall not be liable if it can prove that it was not the one who placed the sign, brand, logo or stamp that identifies it as apparent producer in the defective product or its packaging.
In the case of medicinal products, foods or foodstuffs intended for human consumption, the persons liable shall not be able to invoke the state of scientific and technical knowledge defence set out before in this answer.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
5. Traditional Medicines and OTC Products: Spain
A brief overview of the situation regarding traditional medicines and OTC products in Spain. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
1. What are the regulatory requirements for traditional, herbal, complementary, or alternative medicines and devices?
Plants traditionally used in Spain for medicinal purposes can be sold to the public without the need for a marketing authorization issued by the AEMPS or the European Medicines Agency. These plants must be offered to the consumer without mentioning any properties for treating or preventing diseases.
Besides, herbal medicines, homeopathic medicines, vaccines and medicinal gases are considered medicinal products, and can only be sold to the public if they have a marketing authorization issued by the AEMPS or the European Medicines Agency. Herbal medicines traditionally used in Spain for medicinal purposes, as well as non-injectable homeopathic medicines that are offered to the public without claiming their value in one or more therapeutic indications, can benefit from a simplified authorization procedure.
The marketing of other alternative products, such as food supplements based on vitamins, minerals and other substances, may be subject to the obligation to communicate their placing on the market to the Spanish competent authorities.
2. Can these traditional, herbal, complementary, or alternative products be advertised directly to the public?
Traditional, herbal, complementary or alternative products which are not a medicinal product and/or a medical device may be advertised directly to the public provided the requirements described in Question 3 are met.
3. What health, advertising, and marketing claims may be made for traditional, herbal, complementary, or alternative products?
Plants traditionally used in Spain for medicinal purposes must be offered to the consumer without mentioning any properties for treating or preventing diseases.
Regarding other traditional, herbal, complementary, or alternative products, Royal Decree 1907/1996 states that such products cannot be advertised in Spain if they:
- Claim properties for preventing, treating or curing diseases.
- Claim slimming properties or against obesity.
- Offer relief or healing certainties.
- Claim to have authorizations, homologations or certificates issued by health authorities of any country.
- Claim their use in healthcare centers or their distribution through pharmacies.
- Are accompanied by healthcare professionals, famous people, or real or suspected patients’ testimonials.
- Suggest that their use or consumption enhance physical, mental, sports or sexual performance.
- Use the term “natural” as a characteristic linked to intended preventive or therapeutic effects.
- Attribute a superfluous nature to medicinal products or medical devices or that are presented as an alternative to such products.
- Attribute a superfluous nature to the intervention of healthcare professionals or that are presented as an alternative to the intervention of said professionals.
These prohibitions do not apply to medicinal products and medical devices that are governed by their specific regulations. Likewise, food products may also claim healthy properties and properties for the prevention of diseases under the terms and with the limitations provided in the European Regulation (EC) No 1924/2006 on nutrition and health claims made on foods.
4. What are the regulatory requirements for over-the-counter (non-prescription) medications?
Over the counter (OTC) medicinal products are governed by the rules of Spanish Law on Medicinal Products as well as Royal Decree 1345/2007, which regulates the authorization and registration procedure for medicinal products industrially manufactured.
Advertising to the public of OTC medicinal products which are not reimbursed in Spain is allowed. Advertisements shall include the following claim: “This product is a medicinal product. Refer to the leaflet. If in doubt, consult your pharmacist”.
In addition, such advertisements shall not include any information that:
- leads to the conclusion that no medical appointment or surgical procedure is necessary and that induces a certain diagnosis, or treatment by correspondence;
- suggests that the effect of the medicinal product is guaranteed, with no adverse reactions or side effects, with results greater or equivalent to those of another treatment or medicinal product;
- suggests that the person’s normal health condition may be improved by the use of the medicinal product;
- suggests that the person’s normal health condition may be impaired in case the medicinal product is not used (except for vaccination campaigns approved);
- is exclusively or mainly targeted at children;
- refers to a recommendation from scientists, healthcare professional or other persons, who because of their celebrity may encourage the consumption of medicinal products;
- suggests that the medicinal product is food, a cosmetic or personal hygiene product, or any other consumption product;
- suggests that the safety or efficacy of the medicinal product is due to the fact that it is a natural product;
- may lead to an erroneous self-diagnosis through a detailed description or representation of patient history;
- suggests that its use enhances sports performance;
- refers to alarming, abusing or misleading healing testimonials;
- uses abusively, alarmingly or misleadingly images of alternations of the human body produced by disease injuries, or of the action of the consequences of a medicinal product to the human body; or
- mentions that the medicinal product has obtained the health authority or any other authority.
As regards the advertising of medical devices to the public, the prior authorization from the Spanish regional authorities must be previously obtained. Spanish Law on Medicinal Products expressly prohibits to advertise to the public medical devices which are publicly financed and/or medical devices which are intended to be used or applied exclusively by HCP. Also, according to Royal Decree 1591/2009, it is prohibited to advertise to the public (i) medical devices for self-diagnosis (except for fertility, pregnancy or HIV diagnosis devices), and/or (ii) genetic diagnosis devices.
5. Are there any limitations on locations or channels through which OTC products may be sold?
OTC medicinal products must be sold and delivered by pharmacies only. OTC medical devices may also be sold and delivered to consumers in other stores, such as supermarkets or cosmetic shops. As regards distance selling to the public (e.g. via post) of medicinal products and medical devices, please refer to Chapter 3, Questions 18 and 19 .There are no restrictions in terms of places of sale or distance selling of other OTC products, except for limitations indicated in Question 3.
6. What health, advertising, and marketing claims may be made for OTC products?
Please refer to Chapter 4, Question 3 and 4.
7. Can OTC products be marketed or advertised directly to the public?
Please refer to Question 3 and 4.
8. What is the mechanism by which a prescription-only product can be converted to an OTC product?
Medicinal products shall be subject to medical prescription when: (i) they are likely to present a danger either directly or indirectly, even when used correctly, if utilized without medical supervision, (ii) they are used frequently, and in a very considerable way, under abnormal conditions of use and this may involve, directly or indirectly, a danger to health; (iii) contain substances or preparations based on these substances whose activity or adverse reactions need to be studied in more detail, or (iv) they are administered parenterally, except in exceptional cases, by medical prescription.
The AEMPS may modify the classification granted to a medicinal product (from prescription-only to OTC and vice versa), when new information is obtained that justifies the re-evaluation of the file. When this happens, a period of one year of data exclusivity is granted from the date in which the authorization of modification is granted.
9. What are the requirements for the importation of either traditional medicines or OTC products?
OTC medicinal products may be imported by the marketing authorization holder in Spain or by a legal entity authorized as importer of medicinal products by the AEMPS. In the event that OTC medicinal products come from EU countries, parallel imports requirements must be followed (please refer to Chapter 3, Question7).
Besides, to import OTC medical devices from non-EU countries, a specific authorization from the AEMPS is required. Medical devices acquired from other EU countries can freely circulate in Spain. To import cosmetics, it is necessary the previous presentation of an affidavit before the AEMPS indicating the contact qualified person details.
As regards importation of OTC products different from the above mentioned, there are no specific rules and general rules on importation and intracommunity trade of goods apply.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
6. Marketing, Manufacturing, Packaging & Labeling, Advertising: Spain
All about marketing, manufacturing, packaging & labeling, advertising in Spain. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
1. What is the authorization process for the marketing of new drugs, biologics, medical devices, over-the-counter medications, and other medicinal products?
The AEMPS is the authority in charge of granting marketing authorizations in Spain, whether they result from a national procedure or from a mutual-recognition or decentralized procedure. Marketing authorizations granted by the AEMPS are regulated by Royal Decree 1345/2007. Some provisions of such Royal Decree also affect medicines authorized by the European Commission pursuant to the centralized procedure.
The AEMPS shall grant an authorization if the product:
- fulfils the established quality requirements;
- is safe under normal conditions of use;
- is effective in the therapeutic indications;
- is correctly identified; and
- provides the patient with the necessary information.
The positive therapeutic effects of the medicinal product shall be assessed in relation to any risk for the patient’s health or public health, viewed under a risk-benefit perspective.
The key stages of the procedure are the following:
- submission of the application to the AEMPS;
- validation and acceptance of the submission;
- issuance of the evaluation report; and
- resolution of the application, and issuance, where appropriate, of the marketing authorization of the product.
The maximum period to notify to the applicant the resolution of the authorization procedure is 210 calendar days.
As regards marketing of medical devices, except for custom-made devices, all medical devices must bear the CE marking of conformity when they are placed in the Spanish market (please refer to Chapter 1, Question 3). Likewise, for class IIa, IIb and III devices, a communication must be made to the AEMPS the first time that a person places a medical device in the Spanish market for its distribution or use.
2. What is the authorization process for the marketing of generic versions of these products?
As regards the authorization process for the marketing of generic versions of medicinal products, such process is simpler than the one foreseen for brand-name medicinal products because the applicant shall not be required to provide the results of pre-clinical and clinical trials if it can demonstrate that the medicinal product is a generic medicinal product of a reference medicinal product which is or has been authorized for not less than eight years in a Member State of the European Union or in the Community.
The applicant may also replace the pre-clinical and clinical trial results with appropriate scientific literature, if he can demonstrate that the active substances of the medicinal product have been in well-established medical use within the Community for at least ten years, with recognized efficacy and an acceptable level of safety.
Finally, where the medicinal product possesses the same qualitative and quantitative composition in terms of active substances and the same pharmaceutical form as another medicinal product that has already been authorized, the applicant may rely on the pre-clinical and clinical documentation of the authorized medicinal product, with the permission of the holder thereof.
In the case of biological medicinal products which are similar to a reference biological medicinal product but do not meet the conditions in the definition of generic medicinal products, the results of appropriate pre-clinical tests or clinical trials relating to these conditions must be provided.
3. What are the typical fees for marketing approval?
Please refer to Chapter 1, Question 4.
4. What is the period of authorization and the renewal process?
Please refer to Chapter 1, Question 5.
5. What are the requirements, if any, for post-approval pharmacovigilance?
Royal Decree 577/2013 imposes the following post-approval pharmacovigilance obligations to the marketing authorization holder:
- respecting the Good Practices on Pharmacovigilance for the pharmaceutical industry published by the AEMPS;
- having an adequate pharmacovigilance system, including a master file of the system and undertake periodic audits;
- having a suitably qualified person responsible for pharmacovigilance in the European Union;
- having a contact person for pharmacovigilance in Spain;
- submitting periodic safety reports to the European Medicines Agency;
- having a risk management system for each medicinal product;
- notifying and recording in the Eudravigilance database suspected adverse reactions to the medicinal product;
- monitoring worldwide scientific literature related to the medicinal product;
- carrying out post-authorization studies of efficacy and/or safety required by the competent authorities; and
- performing a continuous evaluation of the benefit-risk parameters of the medicinal product.
Furthermore, products subject to additional monitoring requirements must include a black inverted triangle in their package leaflet and data sheet, accompanied by the phrase “this medicine is subject to additional monitoring”.
6. Are foreign marketing authorizations recognized?
Under Royal Decree 1015/2009, the recognition of a medicinal product that has a foreign marketing authorization requires prior approval from the AEMPS. Approval is subject to the following requirements:
- there is no other adequate medicinal product authorized in Spain;
- a physician must justify in writing the prescription; and
- the patient must consent in writing to the prescription, after having been duly informed.
7. Are parallel imports of medicines or devices allowed?
According to Royal Decree 1785/2000, parallel imports are allowed in Spain, but the following requirements must be met:
- the medicinal product in question needs to have a marketing authorization both in the country of origin as well as in Spain;
- the company responsible for the parallel import must obtain the prior authorization from the AEMPS;
- the labelling and leaflet of the product must comply with the provisions of Royal Decree 1345/2007; and
- the parallel importer must have an authorization as manufacturer of medicinal products in Spain if it carries out in Spain any repackaging and relabeling of the imported product (otherwise, the importer must have an authorization in Spain to perform the activities of a wholesaler).
Intellectual property rights cannot be used to oppose parallel imports. However, before beginning the marketing of a product that has been introduced in Spain through parallel import, the importer must notify the marketing authorization holder of the medicinal product in Spain of its intention to carry out such marketing in Spain (providing, if requested, a sample of the reconditioned product to be marketed by the importer).
8. What are the restrictions on marketing practices such as gifts, sponsorships, consultancy agreements, travel and entertainment, or other incentives for healthcare organizations and individual medical practitioners?
Under the Code of Farmaindustria (Farmaindustria is the Spanish innovative medicinal products industry association) providing gifts to healthcare organizations is subject to certain restrictions. The main restrictions are: that gift must not constitute an inducement to prescribe, recommend, purchase, supply, sell, administer or use any particular product; the gift must be for the internal use of the institution in general and not for the use of an individual; and the provision of the gift must be formalized in writing and the receiving company shall retain a copy of such document.
As regards gifts to healthcare professionals (‘HCP’), the following restrictions contained in Royal Decree 1416/1994 apply: the gift must be relevant for the practice of medicine or pharmacy and the cost of the gift must be insignificant. Moreover, according to the Code of Farmaindustria gifts to HCP are subject -among others- to the following general restrictions: (i) the gift must have stationery or professional use, (ii) it must not be related to a prescription-only medicinal product, and (iii) its market price must not exceed EUR 10. Informational or educational materials and items of medical utility can be given to HCP as a gift subject to certain restrictions: (i) such materials must be directly relevant to the practice of medicine or pharmacy, (ii) they must directly benefit patient care, (iii) they must not affect the routine practice of the HCP, and (iv) their market price must not exceed EUR 60. In addition, the Code of Farmaindustria prohibits the offering or provision of gifts for stationery or professional use within the framework of (i) medical visits mainly related to prescription-only medicines, and (ii) scientific and professional meetings organized by a third party, where promotion mainly pertains to prescription-only medicines, with the exception of pens or pads included in the congress bag, that shall not include any kind of corporate/institutional advertising or product advertising. Within the framework of scientific and professional meetings organized by a company, only pens and pads may be offered or provided; as long as that they are not related to a prescription-only medicine and their market price does not exceed EUR 10 (tax included).
Sponsorship of scientific meetings or congresses for HCP, as well as organizing informative, professional and/or scientific meetings is allowed but subject to restrictions. Such sponsorship must be stated in all documents related to the event, as well as in any published derivative work. It is also possible to pay for the necessary travel, accommodation and enrolment costs to HCP attending such congresses or meetings. According to Royal Decree 1416/1994, such hospitality must be reasonable, meaning that it must not exceed what recipients would normally be prepared to pay for themselves, and such hospitality must remain secondary to the main scientific objective of the event. There are additional restrictions contained in the Code of Farmaindustria, for example: hospitality cannot be extended to accompanying persons, hospitality may be granted only for the duration of the event and one additional day, scientific activities must cover at least 60% of an eight-hour working day, there is a limit of EUR 60 Euros per meal per guest, etc.
As regards entertainment, the hospitality offered to HCP cannot include the organization of social, entertaining or cultural events, except for reasonable welcome cocktails, working meals and gala dinners. The new Code of Farmaindustria, which is in force since 1 January 2021 prohibits any kind of hospitality for training activities or scientific-professional meetings held virtually / by telematic means.
9. How is the manufacturing of medicines and devices regulated and by which agencies?
Performing the industrial manufacturing of medicinal products in Spain requires obtaining an authorization from the AEMPS. The application shall normally be electronically submitted directly to the AEMPS and must be at least in Spanish (although scientific-technical documentation may be submitted in another language). However, non-industrial preparation of compounded medicinal products at hospital and community pharmacies do not require the authorization of the AEMPS.
The key stages in the process to obtain a manufacturing authorization are:
- compilation of the documentation to be submitted to the AEMPS;
- receipt and review of the documentation by the AEMPS;
- compilation and submission of additional information requested by the AEMPS;
- inspection of the manufacturing sites by the AEMPS;
- submission of allegations and additional information as regards objections raised by the AEMPS as a result of the inspection of the manufacturing premises; and
- issuance by the AEMPS of a resolution granting or denying the authorization.
Although the law foresees a period of 90 days for the issuance of the decision regarding the grant of the manufacturing authorization, in practice such decision is issued by the AEMPS around 180 to 270 days after the submission of the application.
The manufacturing of medical devices requires a prior authorization granted by the AEMPS (for custom-made devices an authorization may also be required from the competent regional authorities).
10. Are local manufacturing requirements compatible with Good Manufacturing Practices (GMPs) as defined by the U.S. Food & Drug Administration and/or the European Medicines Agency?
The local manufacturing practices are in accordance with the guidelines issued by the European Medicines Agency.
11. What is the inspection regime for manufacturing facilities?
The health administrations, whenever deemed necessary, are entitled to undertake periodic inspections to the manufacturing facilities. The AEMPS may also inspect the manufacturing facilities whenever it deems necessary, by itself or through the health departments of the relevant Autonomous Regions.
12. Are manufacturing facilities open for inspection by foreign inspectors or third-party inspectors as authorized by the FDA/EMA?
No. The inspections shall be conducted by the AEMPS and any other Spanish competent health administrations. However, there may be inspections undertaken at the request of other agencies, including EMA and FDA.
13. What are the requirements for storage, packaging, and handling of medicines and devices and their constituent components?
The storage, packaging and handling of medicinal products shall comply with the provisions set forth in Royal Decree 824/2010, Royal Decree 782/2013 and the EU Good Distribution Practices of medicinal products for human use.
The storage, packaging and handling of medical devices shall comply with the provisions set forth in Royal Decree 1591/2009.
14. What information must be included in medicine and device labelling?
According to Royal Decree 1345/2007, the labelling of medicinal products must contain the following information:
- name of the medicinal product including the dosage and the pharmaceutical form. Where appropriate, the mention of baby, children or adults. When the product contains up to three active ingredients, the Spanish Official Denomination (DOE) shall be included, otherwise, the International Common Denomination (ICD) or, in its absence, its common name;
- name of the medicinal product in Braille alphabet, considering the particularities of each medicinal product;
- active substances expressed qualitative and quantitative per dosage unit or according to the form of administration for a given volume or weight, using the Spanish DOEs or the ICDs, or, failing that, their common or scientific names;
- list of those excipients known to have a recognized action or effect whose knowledge is necessary for the use of the product and if the product is injectable, or a topical or eye preparation, all excipients must be stated;
- the pharmaceutical form and the contents by weight, volume or number of doses of the product;
- the method and the route of administration;
- a special warning that the medicinal product must be stored out of reach of children;
- any special warning, when the medicinal product requires so;
- In the case of medicinal products containing radionuclides, the dangerous goods transport conditions must be included;
- in case of medicinal gases, the technical specifications they must comply with, the conditions of supply and transport, and, where appropriate, the corresponding symbols must be included;
- expiry date including month and year. In addition, in case of medicinal products with reduced stability after reconstitution, dilution or opening, the shelf life of the reconstituted preparation, diluted or after opening shall be included and they shall also include a box for consignment by users. In case of medicinal products containing radionuclides, the day/ month/year shall be included, and, where appropriate, time: minutes and country of the time reference will be expressed;
- special storage precautions, if any;
- special precautions for disposal of unused medicinal products or waste materials from medicinal products, if appropriate; and if applicable, the symbols authorized by the AEMPS, in order to facilitate the application and development of medicinal products collection systems and to favor the protection of the environment;
- the name and address of the holder of the marketing authorization and, where appropriate, the name of the local representative designated by the holder;
- the National Code of the Medicinal Product;
- the manufacturer’s batch number;
- the number of the authorization for placing the medicinal product on the market;
- in case of medicinal products not subject to medical prescription, the indication of use;
- prescription and dispensation conditions;
- box or blank space to indicate the dosage, duration of treatment and frequency of use or intakes, except in those cases determined by the AEMPS, taking into account the particularities of each medication;
- symbols, acronyms and legends described in Annex IV to Royal Decree 1345/2007; and
- seal of the NHS, when applicable.
According to Royal Decree 1591/2009, the labelling of medical devices shall contain the following information:
- the name or business name and address of the manufacturer. In case of products imported into the EU territory and intended to be distributed in the EU, the label, the name and address of the authorized representative must be included, where the manufacturer does not have a registered office in the EU;
- the information strictly necessary for the user to be able to identify the device and contents of the packaging;
- if applicable, the expression “sterile”;
- if applicable, the batch number;
- if applicable, the deadline for safe use of the device, expressed in the year and month;
- if applicable, indication that the device is for single use;
- if applicable, the expression “custom made device”;
- for devices intended for clinical investigations, the expression “exclusively for clinical investigation”;
- special storage or handling conditions;
- instructions for use;
- any warning and cautions;
- manufacturing year; and
- if applicable, sterilization process.
15. What additional information may be included in labeling and packaging?
Please refer to Questions 14.
16. What items may not be included in labeling and packaging?
Generally speaking, any item that may lead to an irrational use of the medicinal products or medical devices shall not be included in the labelling and or packaging of a medicinal product.
17. What are the restrictions and requirements for the marketing and advertising of medicines and devices?
Advertising of prescription medicinal products or publicly financed medicinal products directed to the general public is strictly prohibited under Royal Decree 1416/1994 and the Code of Farmaindustria. Also, according to Royal Decree 1416/1994, mentioning certain therapeutic indications (i.e. sexually transmitted diseases, other serious infectious diseases, cancer, chronic insomnia, diabetes or other metabolic illnesses) in the advertisement directed to the public of medicinal products is prohibited.
The advertising of non-financed OTC medicinal products must comply with the following requirements: (i) it must clearly indicate that it is an advertisement and that the product advertised is a medicinal product; (ii) it must provide certain information (e.g. the complete name of the product, an invitation to read the instructions of the leaflet and to consult a pharmacist, etc.); and (iii) it must not contain claims or statements that are prohibited (e.g. claims or statements exclusively or mainly targeted at children , claims or statements suggesting that the effect of the medicinal product is guaranteed, with no adverse reactions or side effects, , etc.). Please refer to Chapter 4, Question 4.
According to Royal Decree 1416/1994, advertising of prescription medicinal products is allowed only to HCP. Among the requirements to be complied with, certain information must be provided to the HCP, such as: the dosages and pharmaceutical forms in which the product is available, the prescription and dispensation regime, the retail price and conditions under which the product is publicly financed and the estimated cost of treatment (if possible).
As regards the advertising of medical devices to the public, the prior authorization from the Spanish regional authorities must be previously obtained. On the restrictions, Spanish Law on Medicinal Products expressly prohibits to advertise to the public medical devices which are publicly financed and/or medical devices which are intended to be used or applied exclusively by HCP. Also, according to Royal Decree 1591/2009, it is prohibited to advertise to the public (i) medical devices for self-diagnosis (except for fertility, pregnancy or HIV diagnosis devices), and/or (ii) genetic diagnosis devices.
18. Where can medicines and devices be sold or delivered? Can medicines and devices be sold or delivered via post?
Medicinal products must be dispensed to the public by pharmacies or pharmacy services only. Medical devices may also be sold and delivered to the public by pharmacies. However, there are non-prescription medical devices which may also be sold and delivered to consumers in other stores, such as supermarkets or cosmetic shops.
As regards the distance selling to the public (e.g., via post) of medicinal products and medical devices, there are certain restrictions. According to article 3.5 of Spanish Law on Medicinal Products, it is strictly prohibited to sell medicinal products and medical devices which are subject to prescription via post.
Distance selling of non-prescription medicinal products and medical devices to the public is not entirely regulated in Spain. There is one specific regulation issued in this regard in connection with medicinal products for human use. We refer to Royal Decree 870/2013 regulating the distance selling of non-prescription medicinal products to the public. According to the conditions contained therein, pharmacies are entitled to have their own official website to sell OTC medicinal products to the public.
However, certain Spanish Regions have passed regional regulations which allow other forms of home delivery in exceptional cases (such as home delivery in isolated rural areas or to people who are dependent).
19. What are the restrictions and requirements for electronic marketing and advertising via email, by Internet, social media, and other channels?
The same restrictions and requirements applicable for non-electronic marketing and advertising apply to the electronic marketing and advertising (please refer to Question 17).
20. May medicines and devices be advertised or sold directly to consumers?
Please refer to Questions 17 and 18.
21. How is compliance monitored?
Compliance monitorization is exercised by the Spanish regional authorities via prior authorization and/or control ex-post.
Advertising of medical devices to the public is monitored via prior authorization.
Advertising of medical devices to HCP or advertising of medicinal products to the public or to HCP is monitored via control ex post. Monitorization ex-post means that the authorities can inspect and review the advertising activities carried out by the company to ensure compliance with the applicable legal requirements. In case of advertising materials addressed to HCPs, the company must send a copy of such materials to the competent authority of the Autonomous Region where said company is located at the time of their distribution in order to facilitate the monitoring tasks of these authorities.
22. What are the potential penalties for noncompliance?
Please refer to Chapter 1, Question 9.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
7. Preclinical & Clinical Trial Requirements: Spain
Preclinical & clinical trial requirements in Spain – a comprehensive legal overview. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain, available to purchase here for GBP 119.
1. Are clinical trials required to be conducted locally as a condition (stated or implicit) for marketing approval?
No, clinical trial may be conducted abroad. For those cases whenever clinical trials are conducted outside the European Union, the applicant of the marketing authorization shall submit a declaration stating that the clinical trials carried out outside the European Union meet the ethical requirements set forth in the Spanish legislation.
2. How are clinical trials funded?
Most clinical trials conducted in Spain are sponsored by the pharmaceutical industry, being funded by the sponsoring companies. Clinical trials in Spain might be sponsored also by individuals, institutions or organizations, either private or public.
3. What are the requirements for preclinical and clinical trial protocols? Who must approve the protocols?
The clinical trials protocols shall describe the reasons, objectives, design, methodology, statistical considerations and organization of a clinical trial. The authorizing of clinical trials in Spain by the AEMPS requires prior assessment of the protocol, done jointly, by the AEMPS and the Ethics Committee for research with medicinal products.
4. What are the requirements for consent by participants in clinical trials?
Trial participants must freely give their consent before being included in a clinical trial. Consent must be given after having been informed of all aspects of the clinical trial that are relevant to the participant’s decision to participate. Minors or incapacitated persons’ consent must be given through their legal representatives. Consent must be given in writing. The principal investigator is normally in charge for obtaining informed consent from trial subjects.
The Ethics Committee shall approve the process for obtaining consent from trial subjects and the patient information sheet / informed consent form. Such documents must be at least in Spanish.
5. May participants in clinical trials be compensated?
According to Spanish law participants may only be reimbursed for any expenses or losses related to their participation in trials. However, in special situations, the Ethics Committee may report favourably the compensation to participants for the inconveniences derived from their participation in the clinical trial, provided that said compensation does not influence the decision of the participant to participate in the clinical trial.
6. How are participants in clinical trials protected and indemnified against any harm that arises as a result of participation in the trial?
Participants will be compensated against any personal damage caused as a result of their participation in the clinical trial, as well as the economic damages that derived from said personal damage, provided that this damage is not inherent either: (i) to the pathology under study or (ii) to the natarual course of the disease of the participant as a result of the ineffectiveness of the treatment.
Under Spanish law, one of the conditions for conducting a clinical trial is the contracting, by the sponsor, of a civil liability insurance covering the civil liability the sponsor, the principal investigator and the investigator’s team, and the site against any claim brought by participants for damages suffered due to the clinical trial. The minimum guaranteed amount shall be EUR 250,000 per trial subject. A maximum insured capital per trial and per year of EUR 2,500,000 may be established.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs
Also from this Legal Handbook
8. Regulatory, Pricing and Reimbursement: Spain
Key legal info on regulatory, pricing and reimbursement in Spain. Prepared in association with Faus Moliner Abogados, a leading law firm in Spain, this is an extract from The Pharma Legal Handbook: Spain.
The guide is available to purchase here for GBP 119.
1. What are the regulatory authorities with jurisdiction over drugs, biologicals, and medical devices in your country?
The main regulatory authorities in Spain are:
- The Spanish Ministry of Health (‘Spanish Ministry of Health’), which is the department of the central Spanish government responsible, among others, for drafting and implementing the rules on pricing and reimbursement of medicinal products that are financed through public funds in Spain.
- The Spanish Medicines Agency of Medicinal Products and Medical Devices (i.e. Agencia Española de Medicamentos y Productos Sanitarios – ‘AEMPS’), which is also part of the central Spanish government, is responsible, among others, for granting marketing authorizations for medicinal products in Spain through the national, mutual-recognition, or decentralized procedures foreseen in the European regulations and with jurisdiction over medical devices and cosmetics as well.
2. What is the regulatory framework for the authorization, pricing, and reimbursement of drugs, biologicals, and medical devices?
The Spanish current regulatory framework regarding authorization, pricing and reimbursement of medicinal products, including biologicals, and medical devices includes (i) Royal Decree-Legislative 1/2015, approving the revised text of the Law on Guarantees and Rational Use of Medicinal Products and Medical Devices (‘Spanish Law on Medicinal Products’), (ii) Royal Decree 1345/2007 on authorization of industrially manufactured medicinal products, (iii) Royal Decree 271/1990, Royal Decree 83/1993, Order of 17 December 1990 and Order of 6 April 1993 on price and reimbursement, and (iv) Royal Decree 177/2014 establishing the Reference Price System. Likewise, Royal Decree-Law 8/2010 (amended by Royal Decree-Law 9/2011), established different mandatory discounts on the ex-factory price of the medicinal product when such medicinal product is dispensed or administered to patients through public funding.
The Spanish current regulatory framework regarding authorization, pricing and reimbursement of medical devices includes (i) Spanish Law on Medicinal Products, (ii) Royal Decree 1591/2009, on medical devices, (iii) Royal Decree 1616/2009, on active implant medical devices, and (iv) Royal Decree 1662/2000, on “in vitro” diagnostic medical devices.
3. What are the steps to obtain authorization to develop, test, and market a product?
Any person or entity that wishes to manufacture medicinal products or medical devices in Spain, must be previously authorized as manufacturer by the AEMPS, in compliance with the provisions contained in Royal Decree 824/2010 or Royal Decree 1591/2009, as the case may be.
A medicinal product can be placed in the Spanish market if it previously holds a marketing authorization obtained by one of the following procedures:
- national procedure, mutual-recognition procedure, or decentralized procedure at the AEMPS; or
- centralized procedure at the European Medicines Agency (‘EMA’).
As regards medical devices, please note that they are divided in four classes (III, IIb, IIa and I), ranked mainly considering the level of invasiveness of the device, the part of the body it is in contact with and the duration of such contact. Except for custom-made devices, all medical devices must bear a CE marking of conformity when they are placed in the market in Spain. The CE marking evidences conformity of the device with the requirements of the applicable laws. For class I devices, such conformity shall be evaluated and declared under the exclusive responsibility of the manufacturer. For class IIa, IIb and III devices, the declaration of conformity requires an evaluation of the device by a notified body (the AEMPS in Spain or any other notified body of another European Union member state). Additionally, for class IIa, IIb and III devices, a communication must be made to the AEMPS the first time that a person places a medical device for distribution or use in the Spanish market.
Furthermore, testing of medicinal products or medical devices must be carried out in accordance with clinical investigation rules, which are mainly contained in Royal Decree 1090/2015 regulating clinical trials.
4. What are the approximate fees for each authorization?
Fees for services provided by the AEMPS are approved on an annual basis. Current fees were approved by Law 22/2021 on the budget for 2022. A list of the services provided by the AEMPS and the corresponding fees may be found at: https://www.aemps.gob.es/industria-farmaceutica/tasas/relaciontasas/?lang=en
AEMPS’s services/fees include, among others, the following:
- Fee for the evaluation, authorisation and registration of a new medicine for human use (no generic): 21,576.3 Eur
- Fee for the evaluation, authorisation and registration of a new generic medicine for human use: 8,776.68 Eur
- Authorisation of a new pharmaceutical laboratory: 6.156,58 Eur
- Authorisation and/or certification for medicines warehouses under customs control or supervision: 1,366,31 Eur
- Individual inspection activities in Spain, unless a complaint has been made or it is requested by a representative association of consumers: 5,208.19 Eur
5. For how long are marketing authorizations/registrations valid? How are marketing authorizations/registrations renewed?
The marketing authorization of a medicinal product is valid for an initial period of five years. At least nine months before expiration of period, the marketing authorization holder may apply for a marketing authorization renewal, pursuant to article 27 of Royal Decree 1345/2007. Together with the renewal application, the applicant must pay the relevant fees (EUR 2,437.84) and provide a consolidated version of the registration dossier including evaluation data contained in suspected adverse reactions reports and periodic safety update reports, as well as all the variations introduced since the marketing authorization was granted.
Once renewed, the marketing authorization will be valid for an unlimited period, unless the AEMPS requires an additional five-year renewal based on duly justified pharmacovigilance-related reasons.
6. How does the authorization process differ between brand-name products and generic products? Are there differences for local manufacturers versus foreign-owned manufacturers?
The authorization of both a brand-name medicinal product and a generic medicinal product follow the same legal process. There are only minor differences as regards the fees to be paid to the AEMPS (as explained in Chapter 1, Question 4) and as regards the application documentation since the authorization procedure of a generic medicinal product does not require the applicant to provide pre-clinical tests and clinical trials results (bioequivalence studies must be provided instead of).
The law does not contemplate any difference for local manufacturers vs foreign-owned manufacturers when it comes to the authorization procedure for medicinal products.
7. How are combination products (drug + drug, drug + biologic, drug + device, biologic + device, drug +biologic + device) regulated?
There is no specific regulation for combination products in Spain. Please note that biologics are considered as medicinal products and, thus, subject to the same rules.
A combination product comprising both a medicinal product and medical device may be authorized as a medicinal product, e.g., an injectable medicinal product that comes in a pre-filled syringe; such product may be authorized as a medicinal product (although the syringe itself may be authorized as a medical device). It is also possible that a combination product comprising both a medicinal product and medical device is authorized as medical device, e.g. a medical device incorporating as an integral part a substance which, used separately, may be considered as a medicinal product (Judgement of the European Court of Justice in the Case C-527/17 Boston Scientific v Deutsches Patent).
The AEMPS has the final decision on the classification of any such combination product.
8. How is compliance with regulation monitored and evaluated? Is the regulatory regime comparable with the U.S. Food and Drug Administration or the European Medicines Agency expectations and requirements?
The AEMPS as well as the Health Authorities of the Autonomous Regions (Spain is divided in 17 Autonomous Regions) are in charge of monitoring compliance with the applicable regulations in Spain. As the regulatory regime applicable in Spain is based in EU legislation and guidelines, it is aligned with the European Medicines Agency expectations and requirements.
9. What is the potential range of penalties for noncompliance?
The potential penalties for noncompliance of the regulatory regime range from EUR 30,000 to EUR 1,000,000. The amount of the penalty may exceed up to five times the value of the products or services subject to the infringement in case of very serious infringements. Examples of noncompliance include elaborating, manufacturing, importing, exporting, dispensing or distributing medicines without authorization to do so; modifying any of the conditions of the authorization without having the consent of the authorities; or marketing medicinal products without having obtained the authorization to do so.
Additional sanctions may also be agreed, such as confiscation of the illicit profit that has been obtained or shutting down the activities of the company during a period up to five years, in case of very serious infringements (Article 114.2 of the Spanish Law on Medicinal Products). Furthermore, sanctions for the commission of serious and very serious infringements will be published in the corresponding official journal.
10. Is there a national healthcare system? If so, how is it administered and funded?
Yes, there is a national healthcare system. The NHS is administered by the Spanish Ministry of Health and also by the Health Departments of the Autonomous Regions. It is funded by tax of the contributors. The Ministry of Health is the department of the Spanish central government responsible for approving pricing and reimbursement of medicinal products. However, since the public funds that may be used to finance the reimbursement of medicines come out of the budget of the 17 Autonomous Regions in which Spain is divided, such regions participate in the committee of the Ministry of Health responsible for assessing applications for price and reimbursement of medicinal products (the “CIPM”).
11. How does the government (or public) healthcare system function with private sector healthcare?
Hospitals belonging to the National Health System (‘NHS’) are in the public sector, under the jurisdiction of the Spanish Ministry of Health as well as under the jurisdiction of the Health Authorities of the Autonomous Regions. Private sector hospitals, both profit-making and non-profit-making, have their own management arrangements.
The Ministry of Health and regional health administrations may contract with private hospitals to provide healthcare services to the users of the NHS whenever this is advantageous, especially considering the quality-cost binomial, and provided that the right of access is guaranteed.
12. Are prices of drugs and devices regulated and, if so, how?
Prices of medicinal products that are reimbursed may not be freely set by the marketer as they require the prior approval from the Ministry of Health/CIPM. Also, in case that there are legitimate public reasons, the Ministry of Health/CIPM may control the price of medicinal products that are excluded from the reimbursement.
Royal Decree 271/1990 states that the maximum ex-factory price of a reimbursed medicinal product should be equal to the cost of the product plus a given margin (12 – 18 per cent on capitals allocated to exploitation). As a matter of practice, however, the process of setting the price of a reimbursed medicinal product entails a negotiation of the price with public authorities. Additionally, companies are legally obliged to grant a discount on the maximum ex-factory price approved by the authorities. This is regulated in Royal Decree-Law 8/2010.
Prices of reimbursed medical devices dispensed to non-hospitalized patients are also regulated (see Spanish Law on Medicinal Products and Royal Decree 9/1996) and fixed by the Ministry of Health/CIPM.
13. How are drugs and devices used by patients paid for? What roles do public and private payers play?
Autonomous Regions are the ones who pay for all healthcare services, including drugs and devices, out from their own budgets, and, subject to certain conditions which may derive from European and Spanish rules on public procurement, they enjoy a large degree of autonomy to decide how they purchase goods and services which they may require in order to provide healthcare services to patients.
The NHS pays for all those drugs and devices which are dispensed in hospitals. On the other hand, products that patients obtain at retail pharmacies are subject to copayment rules under which the patient has to pay part of the price of the product. The copayment percentage depends on the type of product and also on the type of patient.
14. Who dispenses drugs and devices to patients and how are those dispensers compensated?
Medicinal products can only be dispensed by the pharmacists. The patient withdraws the medicinal product from the pharmacy and, if applicable, pays to the pharmacist the amount of the copayment established by the law. Subsequently, pharmacy invoices the government of the Autonomous Region where it is established the selling price of the medicinal product (the maximum ex-factory price approved by the Ministry of Health), plus the margin set forth in the law for the wholesaler and for the pharmacy, less the amount paid by the patient. In the case of the medicinal products that are administered to patients in public healthcare centres and hospitals, such products are not paid by the patients but financed from the budget of the centre itself.
15. What are the professional and legal responsibilities of those who dispense drugs and devices? What role do they play in providing patient care, information, and safety?
Pharmacists, as responsible for dispensing drugs and devices to the citizens, will ensure compliance of the guidelines established by the physician responsible for the patient in the prescription and will cooperate in the follow-up of the treatment by checking their effectiveness and safety.
Likewise, they will participate in activities aimed at the rational use of drugs, in particular through the information they provide to the patient. Once the drug has been dispensed, they will be able to provide personalized dosing systems to patients who request it in order to improve adherence to the treatments established by the competent health authorities. The conditions and requirements of such personalized dosing systems shall also be established by health authorities.
Click the following links to read more legal articles from Spain:
- Regulatory Pricing and Reimbursement Overview
- Preclinical and Clinical Trial Requirements
- Marketing, Manufacturing, Packaging & Labeling, Advertising
- Traditional Medicines and OTC Products
- Product Liability
- Patents & Trademarks
- Regulatory Reform
- Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs