The Pharma Legal Handbook: Brazil





 Brazil is home to a USD 14.92 billion pharma market that represents around two percent of the global total, grew by 14.21 percent in 2021, is the largest in Latin America and the eighth biggest globally.
Brazil is home to a USD 14.92 billion pharma market that represents around two percent of the global total, grew by 14.21 percent in 2021, is the largest in Latin America and the eighth biggest globally.
Drawn in by these outstanding fundamentals, increasing numbers of international investors are now looking toward the Brazilian market. For anyone hoping to capitalise on the abundant opportunities in Brazilian pharma, The Pharma Legal Handbook: Brazil will be an invaluable resource.
Prepared in association with Trench Rossi e Watanabe, a leading Brazilian law firm, it should answer any questions linked to regulation, pricing, clinical and preclinical trials, marketing, manufacturing, trademarks and patents.
May 2022
1. Biosimilars & Biologics: Brazil
Biosimilars & biologics in Brazil – a comprehensive legal overview. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. Are biosimilar medicines considered the same as generic medicines in your country?
No, a biosimilar drug is not considered to be the same as a generic drug. There are specific rules governing generic and similar drugs, and they are solely applicable to synthetic drugs, and not to biologicals, which are ruled by a specific resolution.
For biological products, ANVISA’s Resolution RDC No. 55/2010 determines that there are two types of products: “new biological products” (for products not registered before ANVISA yet) and “biological products” (for the products that are not new or that contain an already known molecule of biological nature).
Note, however, that the rules establish two different routes to register “biological products”: by comparison, which, by definition, “is the scientific comparison, with respect to non-clinical and clinical parameters in terms of quality, efficacy and safety, of a biological product with a comparator biological product, in order to establish that there are no detectable differences in terms of quality, effectiveness and safety between the products” and by individual pathway, defined as “the regulatory route that may be used for obtaining the registration of a biological product in which it is necessary to present total data on development, production, quality control and non-clinical and clinical data to demonstrate quality , efficacy and safety of the product”.
Thus, despite the fact that Brazilian legislation does not adopt the wording “biosimilar”, but only “new biological products” and “biological products” (for the products that are not new or that contain an already known molecule of biological nature), “biosimilar” is the “biological product” registered by comparison pathway.
2. Are all biologic medicines, including biosimilar medicines patentable in your country?
Biologic and biosimilar medicines are generally patentable in Brazil, provide they comply with patentability requirements, such as novelty, inventive step and industrial application.
Federal Law No. 9.279/1996 (Brazilian IP Law) also establishes, however, that biological materials, when found in nature or isolated therefrom, are not considered inventions (Article 10, IX, of the Brazilian IP Law). For this reason, biological materials such as nucleic acids, antibodies and vectors can only be patented if they comprise human-induced modification(s) in their genetic composition.
In addition, natural living beings (animals and plants), in whole or in part (such as cells), even if modified/recombinant ones, cannot be patented under Article 18, III, of the Brazilian IP Law. However, this does not exclude the possibility of having transgenic microorganisms, such as bacteria, protozoa, virus, and yeasts protected by a patent. For the purposes of the IP Law, transgenic micro-organisms are organisms, except the whole or part of plants or animals, that exhibit, due to direct human intervention in their genetic composition, a characteristic that cannot normally be attained by the species under natural conditions.
3. Is there a specific regulatory framework for the marketing authorization of biosimilar medicines in your country? If yes, what is the regulatory framework for the authorization of biosimilar medicines?
Yes. ANVISA’s Resolution RDC No. 55/2010 is the main rule governing biologicals.
4. What kind of data package is needed to obtain approval for a biosimilar drug? Is this any different to the requirements for the original Biologics drug?
To obtain approval for a biologic drug (new biological product), it is necessary to submit to ANVISA documents about the company (such as the Sanitary Authorization and the Certificate of Technic Responsibility) and about the drug (such as its pharmacovigilance data, identification code and specifications). It is also necessary to submit in the same proceeding a technical report, a therapeutic experimentation report and a pharmacovigilance report in order to detail every methodological, clinical, operational and functional aspect of the biologic drug. If the product is blood derivative, biotechnological or if it is a vaccine, certain specific documents are also required.
In reference of the biosimilar drugs (biological products), the requirements follow the original biologic drug pattern, except as follows:
For biological products registered by individual pathway: The dossier required is the same aforementioned, but, in this case, the extension of the non-clinical studies can be reduced, depending on the complexity of the molecule. In addition, the clinical studies in its phases II and I are not mandatorily comparatives. Studies phase III are always necessary (and comparative). If available, the results of step IV must be submitted.
For biological products registered by comparison pathway: The dossier required is the same as aforementioned, with the addition of a data report about the biologic product and its comparator.
5. What are the requirements for the choice of the reference comparator product?
The comparator product is defined as the already registered biological product, based on the submission of a full dossier, and which has already been commercialized in Brazil.
The comparative pathway may be used when comparability between the biological product/biosimilar and the already registered biological drug is possible with regards to quality, efficacy, and safety. This pathway will require the submission of a comparative dossier containing non-clinical and clinical studies in order to demonstrate comparability between the biological product to be approved and the comparator one. It also requires the submission of studies with information about development and quality control and a comparability result report (providing a comparison to the comparator drug), among other requirements.
6. Can the comparator product be sourced from another regulatory jurisdiction? If yes, what are the data needed to support this approach?
No, the comparator product must be registered before ANVISA.
7. How are the prices of biosimilar medicines regulated? Is this any different to the requirements for the original Biologics drug?
The prices of biological products are regulated by Brazilian Drug Market Regulation Chamber (“CMED”), through the specific parameters defined in Communication No. 9/2016.
The prices applicable to the drugs that evidence therapeutic gain, the price cannot be higher than the lowest price practiced for the same product in United States, New Zealand, Australia, Greece, Portugal, Italy, Spain, France, and Canada.
For drugs that do not evidence therapeutic gain and that are new to company’s portfolio, the price will be defined based on the average cost of treatment with drugs with a similar molecule, considered revenues, and cannot, in any case, be higher than the lowest price practiced for the same product in United States, New Zealand, Australia, Greece, Portugal, Italy, Spain, France, and Canada.
For drugs that do not evidence therapeutic gain but in case the company already has in its portfolio of commercialized products a product with similar molecule, the price will be defined based on the average cost of treatment with drugs marketed by the company itself.
For new presentations of drugs already marketed by the company with the same trademark, the price will be defined based on the average cost of treatment with the same drug.
8. What is the reimbursement policy for biosimilar medicine? Is this any different to the requirements for the original Biologics drug?
Yes, the reimbursement is possible but there is no specific policy. Note, however, that according to court precedents on Federal Law No. 8,080/1990, known as the Brazilian Organic Health Law, the payment or reimbursement of national or imported drugs that are not registered before ANVISA is prohibited. There is no difference to the requirements for new biological products and biological products.
9. Does biosimilar competition impact the reimbursement policy of the originator reference products?
No.
10. What is the legal framework for biosimilar medicines prescribing (clinical decision maker) and dispensing (pharmacy level, hospital or retail)? Is this any different to the requirements for the original Biologics drug?
There is no legal framework specific for prescribing and dispensing of biological products. The physician is free to prescribe the treatment he/she considers appropriate to the patient.
11. Is the system considering physician-led switching and/or pharmacy-level substitution (without involvement of the clinical decision maker)?
The physician is free to change the treatment of his/her patient, since duly justified. However, pharmacy cannot change patients’ prescription.
ANVISA issued a note regarding interchangeability and it considered the evaluation regarding the possibility of switch must be done by the involved physician, on a case-by-case basis, considering the patient and its particularities.
12. What are the post – authorisation requirements (including pharmacovigilance, risk management plans, post-approval studies) for biosimilar medicines? Is this any different to the requirements for the original Biologics drug?
Companies must maintain a pharmacovigilance system in order to ensure the monitoring and evaluation of the safety and efficacy of the drug. There are no differences regarding the requirements for new biological products and biological products.
ANVISA issued a note regarding interchangeability of biologicals and it considered that, although the medical evaluation if mandatory, multiple switches between biological products and the comparator biological product are not adequate, as it jeopardizes the traceability and use monitoring.
13. Are there specific policies and requirements in terms of biosimilar medicines labelling in the event of second medical use patents? Is this any different to the requirements for the original Biologics drug?
No, there are no specific policies.
14. Have there been any significant legal/judicial developments in relation to biosimilars in your country? (Including but not limited to IP, procurement, competition, misleading information campaign, access to reference comparator product)
ANVISA updated in 2018 a note that was initially issued in 2017 regarding interchangeability of biologicals. ANVISA considered that patient’s medical evaluation is mandatory and that the physician is responsible for deciding which products is appropriate to each situation, according to product’s characteristics, existent pathology, individual response and each patient’s treatment history. In view of this, ANVISA expressed that it is not considering classifying biological products as interchangeable or not.
15. Are there proposals for reform or significant change to the legal, regulatory, procurement of biosimilars? If yes, when are they likely to come into force?
The only Bill we identified in relation to biological products is Bill No. 5415/2019 and its purpose is to include new articles to the Federal Law No. 8,080/1990 for prohibiting the dispensation of biological products in the Unified Health System (“SUS”) to a patient that is in ongoing treatment with the new biological product without previous consultation to patient’s physician.
As Bill’s proposition is very recent (dates from October, 2019), it is not possible to estimate if and when it will be approved.
Also from this Legal Handbook
2. Localization: Brazil
Want to know more about localization in Brazil? Read on! Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. Are there any rules or regulations requiring and/or encouraging localization in your country? What is the legal framework defining these localization rules and policies?
Yes, there are rules encouraging localization in Brazil, such as the Federal Law No. 8,666/1993, which is still in force and effect, concurrently with Federal Law No. 14,133/2021 (both provide for Public Procurement rules), and the Federal Decree No. 7,713/2012, which provides for margins of preference of national companies, products and services in the context of acquisition by public entities.
From a regulatory perspective, although the existent rules are not explicit for encouraging localization, only duly licensed Brazilian legal entities are allowed to hold marketing authorizations before the National Health of Surveillance Agency (ANVISA). Therefore, imported health products may only enter into Brazilian market through a Brazilian importer.
Also, the intention of stimulating the local production of drugs and devices, and of enabling Brazilian public laboratories to produce in Brazil involve Brazilian Productive Development Partnerships (PDPs). The PDPs are complex projects whereby a pharmaceutical company undertakes to transfer the technology for the production of a certain drug to a Brazilian state laboratory in exchange for an exclusivity of supply of such drug to the Government market for the period the technology is being transferred. Such exclusivity is materialized by a waiver of a public tender, which is an exception under Brazilian Administrative Law, once the need of a public tender is the general rule under the Constitution and the Public Procurement Law (both Law No. 8.666, of 1993 and Law No. 14,133/2021).
Finally, in 2016 the Federal Law No. 13,243 (Innovation Law) was issued for remodelling and renewing the framework of scientific development, research, and the development of capabilities in science, technology and innovation. In this sense, the Brazilian Government must promote and incentive the research and development of innovative products, services and processes in Brazilian legal entities through the grant of financial, human, material or infrastructure resources.
Specifically regarding the health sector and in the context of the Innovation Law, in 2017 the Federal Decree No. 9,245 was issued for defining the National Policy of Health Technological Innovation (the PDPs, for example, are one of the instruments of such National Policy).
2. Have there been any recent significant changes involving localization rules? If yes, when did they take place and what did they involve?
There have not been any recent significant changes involving localization rules.
3. Is the process of obtaining a marketing authorization impacted by localization policies in your country? If yes, how so (what are the incentives received or the requirements)?
Yes. Marketing authorizations (and sanitary permits) will only be granted by Brazilian authorities (i.e, ANVISA and local health authorities) to Brazilian legal entities. But there is no differential treatment for the place of origin, whether local or external.
4. Is the pricing process for pharmaceutical products impacted by localization policies in your country? If yes, how so (what are the incentives received or the requirements)?
The pricing process for pharmaceutical products is not impacted by localization policies.
5. Is the reimbursement of pharmaceutical products impacted by localization policies in your country? If yes, how so (what are the incentives received or the requirements)?
The reimbursement of pharmaceutical products is not impacted by localization policies.
6. Is the access to public or public tenders of pharmaceutical products impacted by localization policies in your country? If yes, how so (what are the incentives received or the requirements)?
The access to public is not impacted. However, public tenders are.
The Federal Law No. 8,666/1993, Law No. 14,133/2021 and the Federal Decree No. 7,713/2012 provide for margins of preference of national companies, products and services in the context of acquisition by public entities. Specifically regarding the pharma sector, the Federal Decree No. 7,713/2012 sets forth a margin of preference for the acquisition of certain drugs and pharma products (defined in the Decree) in public tenders conducted by the federal government, with the purpose of stimulating the national sustainable development.
The preference margin will be applied only for products manufactured in Brazil, according to certain Brazilian origin rules.
7. Are import tariffs, importation and/or exportation permits, trade and/or taxation of pharmaceutical products impacted by localization policies in your country? If yes, how so?
As a rule, no. However, preferential tariffs may apply upon import and export of goods in case the products originated from a country with which Brazil has a Free Trade or Complementary Economic Agreement. The origin of the product is determined based on the criteria provided in the Agreement, but usually it is connected to the place where the product was subject to the last “substantial transformation”.
8. Are there any other incentives or advantages offered by the current local localization rules in your country? If yes, what are they?
Yes. For instance, Brazil has a Free Trade Zone located in Manaus, State of Amazonas, which was designed to encourage manufacturing for export and local sales. Raw materials, parts and components imported or remitted to the Manaus Free Trade Zone enjoy different tax benefits, including tax deferral and exemption. Other specific tax incentives may apply depending on the specific circumstances of the case, such as the type of product, the purpose of the transaction and the location (State/Municipality) where the transaction is carried out in Brazil.
9. Are there discussions about the possibility of implementing localization policies in your country? If yes, what are the proposed reforms and when should they come into place?
There is currently no discussion as to new localization policies and we do not consider that there would be any changes to these in the near future. In fact, the current government has shown to be adverse to local content policies.
Also from this Legal Handbook
3. Orphan Drugs & Rare Diseases: Brazil
The key facts about orphan drugs & rare diseases in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. What is the definition of Rare Diseases in your country?
According to Resolution RDC No. 205/2017 issued by the National Health of Surveillance Agency (“ANVISA”), “rare diseases” are defined as diseases with incidence of up to 65 cases for every 100,000 inhabitants.
2. Does the designation of ‘Orphan Drug’ exist in your country? (Does it correspond with the definition of Rare Diseases?)
No, there is no specific definition for “Orphan Drug” in Brazilian current rules. However, we understand that it corresponds with the definition of Rare Diseases.
3. What is the regulatory framework for the authorization of an Orphan Drug? (Is this regulatory framework based on Rare Disease status or can it alternatively be based on Orphan Drug foreign status?)
The main applicable rule is ANVISA Resolution RDC No. 205/2017, which provides for a special procedure for approval of clinical trials, good manufacturing practices certification and registration of new drugs for rare diseases.
The conditions for the application of this regulation are: (i) the drug is intended for the treatment, diagnosis, or prevention of a rare disease; (ii) the drug is used in a serious and debilitating condition; and (iii) it proposes to change, in a clinically substantial way, the development of the disease or allows a disease’s remission.
4. Does your country have provisions for relaxed clinical trial/scientific evidence requirements in respect of Orphan Drugs as compared to other drugs?
Yes. Aimed at encouraging companies to request ANVISA’s approval for the conduction of clinical trials in Brazil with experimental drugs for rare diseases, and at making the registration of drugs for rare disease faster, Resolution RDC No. 205/2017 provides for different procedures and criteria when compared to other conventional procedures.
As an example, it allows the company to propose the designation of a drug to treat a rare disease, with ANVISA responsible for the validation stage. In addition, the interested company and ANVISA will meet prior to the submission of the registration application, in order to align about the product and related documents. Also, the rule provides alternatives for cases in which the company applying for registration has not yet completed the entire process of developing a new drug for rare disease.
Also, ANVISA created a new category of products called “advanced therapy medicinal products”, a special category of new drugs comprising the advanced cell therapy product, the tissue engineering product and the gene therapy product, through Resolution RDC No. 505/2021. According to the Resolution, the advanced therapy medicinal products intended for rare diseases will have its registration request analysed with priority.
5. Is there an expedited pathway for Orphan Drugs?
According to information disclosed by ANVISA, the registration pathway related to drugs intended for rare diseases is three times faster than the regular procedure, due to Resolution RDC No. 205/2017. According to Resolution RDC No. 505/2021, certain petitions may be prioritized, such as the ones related to drugs intended for rare diseases (among others).
6. Are foreign marketing authorizations recognized in your jurisdiction for Orphan Drugs? If yes, marketing authorizations from which countries are recognized?
No. It is mandatory to obtain a registration for Brazil, to be granted by ANVISA.
7. Can Orphan Drugs be reimbursed? If so, is there a specific reimbursement procedure for Orphan drugs?
Yes, the reimbursement is possible but there is no specific procedure. Note, however, that according to court precedents on Federal Law No. 8,080/1990, known as the Brazilian Organic Health Law, the payment or reimbursement of national or imported drugs that are not registered before ANVISA is prohibited.
8. How are the prices of Orphan Drugs regulated?
The prices of Orphan Drugs are regulated by Brazilian Drug Market Regulation Chamber (“CMED”). The authority sets limits on drug prices, adopts rules that stimulate competition in the industry, monitors marketing, and enforces penalties when its rules are trespassed.
The company must submit to CMED the request for price approval, after gaining marketing authorization from ANVISA. In order to determine the maximum product price, companies must submit economic data on the product and propose a suggested price. External reference pricing is used to define the price of new drugs.
9. In case of reference price based on a basket of countries, what countries are included?
The prices in Brazil cannot exceed the lowest price charged between nine different countries, which are the United States, New Zealand, Australia, Greece, Portugal, Italy, Spain, France, and Canada.
10. Have there been any significant legal/judicial developments in relation to Orphan Drugs in your country?
On May 2019, Brazilian Federal Supreme Court rendered a decision of general repercussion nature for defining that (i) the State cannot be obliged to provide experimental drugs; (ii) the absence of registration with ANVISA prevents, as a general rule, the supply of drug by judicial decision; (iii) it is possible, exceptionally, the judicial grant of an unregistered drug, in case of unreasonable delay of ANVISA in considering the request (deadline longer than provided for in Law No. 13.411 / 2016), when three requirements are met: (a) the existence of application for registration of the drug in Brazil (except in the case of orphan drugs intended for rare and ultra-rare diseases), (b) the registration of the drug with renowned regulatory agencies abroad, and (c) the lack of a therapeutic substitute registered in Brazil.
11. Are there proposals for reform or significant change to the regulation of Orphan Drugs? If yes, when are they likely to come into force?
As the Resolution RDC No. 205/2017 was recently issued by ANVISA, we do not expect any changes to it in the near future. Note, however, that certain Bills related to orphan drugs are ongoing, such as:
- Bill No. 2233/19: grants tax benefits for drugs intended to rare diseases. In addition, the Bill provides for a simplified registration process for orphan drugs.
- Bill No. 2036/19: creates the temporary special registration of drugs aimed at treating diseases without effective alternatives in the country.
- Bill No. 2.657/2015: provides for the registration and importation, by individuals, of orphan drugs, provides for different criteria for the evaluation and incorporation of orphan drugs and also specifies process for price comparison.
- Bill No. 1606/2011: aims at setting a new national policy on rare diseases.
As the processes are ongoing, we cannot estimate if and when such Bills will come into force.
Also from this Legal Handbook
4. Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs: Brazil
All about cannabinoid drugs, medicinal cannabis and opioid drugs in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
Cannabinoid Drugs
1. Are Cannabinoid Drugs authorized in your country?
The National Health Surveillance Agency (“ANVISA”) recently approved (on December 11, 2019) the Resolution RDC No. 327/19 defining the procedure for granting the Sanitary Authorization for manufacturing and importing Cannabis Products (given name for industrialized products that contain plant derivatives or Cannabis Sativa active agents, with a medicinal purpose). Such rule also establishes requirements for its commercialization, prescription, dispensation, monitoring and supervision.
The aforementioned Resolution provides that the commercialization must occur upon medical prescription and exclusively in pharmacies without manipulation. Furthermore, the rule determines that Cannabis Products must have predominantly Cannabinoid (CBD), and must observe a limit of 0.2% tetrahydrocannabinol (THC), except in cases of palliative care exclusively for patients without other therapeutic alternatives, and in irreversible or terminal clinical situations.
Nonetheless, in order to manufacture and commercialize Cannabis Products in national territory, the interested company shall import the pharmaceutical input into the forms of vegetable derivative, phytopharmaceutical, in bulk, or industrialized product, once planting is still forbidden in Brazil.
Before such rule, only the importation of Cannabis’ based products was possible (through individuals for personal use or by companies interested in registering the product before ANVISA). The manufacturing of the product locally was ruled.
2. What are the regulatory authorities with jurisdiction over Cannabinoid Drugs?
The regulatory authority with jurisdiction over Cannabinoid Products is ANVISA.
Federal Police, Civil Police and Army also have jurisdiction over Cannabinoids, considering that it is a controlled substance.
3. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Cannabinoid Drugs?
The main regulation on these subjects is the aforementioned Resolution RDC No. 327/19, which provides the procedure for granting the Sanitary Authorization (which allows the manufacture and importation of the Cannabis Products), and its commercialization under the terms of the Resolution.
Also, Ordinance No. 344/1998, which determines the substances and drugs subject to special control and Resolution RDC No. 17/2015, which authorizes the importation and determines the procedure for importation by individuals of Cannabis based products.
4. Which are the cannabinoid drugs that have received market approval to date?
Resolution 327/2019 was published on December 11, 2019, thus there is no Cannabis Product approved based in this Resolution.
However, there is a product that received ANVISA’s marketing authorization few years ago based on the general rule for registration of synthetic drugs, the cannabis-based product called Mevatyl (also called Sativex in other countries).
5. Who can prescribe Cannabinoid Drugs?
According to Resolution No. 327/19, prescription is restricted to medical professionals duly registered before the Federal Medical Council, only when there is no other therapeutic option available in the Brazilian market.
6. Is there a list of doctors authorized to prescribe Cannabinoid Drugs?
No.
7. What approvals or notifications are required to prescribe Cannabinoid Drugs?
As mentioned above, the medical professionals must be duly registered before Federal Medical Council.
8. Which organizations are authorized to sell/distribute Cannabinoid Drugs available?
According to Resolution No. 327/19 the commercialization of Cannabis Products must occur exclusively by pharmacies without manipulation, upon presentation of prescription by a medical professional, legally qualified. Such pharmacies must be previously licensed before ANVISA and before local health authorities.
9. Is there a list of retailers/distributors authorized to sell Cannabinoid Drugs?
No.
10. Are there proposals for reform or significant change to the regulation of Cannabinoid Drugs?
The Resolution No. 327/19 was recently approved by ANVISA and will be reviewed after three years of its publication.
11. When are they likely to come into force?
The Resolution No. 327/19 will come into force 90 (ninety) days from the date of its publication (i.e. March 11, 2020).
Medicinal Cannabis
12. Is Medicinal Cannabis authorized in the country?
According to Federal Law No. 11.343/2006, which established the Brazilian ‘National System for Public Policies on Drugs’ (SISNAD), it is forbidden in all national territory planting, growing, harvesting and exploiting vegetables and substrates from which drugs may be extracted or produced. Besides, Cannabis sativa L. is currently considered a proscribed substance by ANVISA on its Ordinance No. 344/1998 and, therefore, Medicinal Cannabis is still forbidden in Brazil.
Its legal use is restricted to the manufacturing and commercializing of Cannabis Products by legally registered companies. These companies, in order to fabricate the drugs, have to import the pharmaceutical input into the forms of vegetable derivative, phytopharmaceutical, in bulk, or industrialized product.
13. What are the regulatory authorities with jurisdiction over Medicinal Cannabis?
The regulatory authority with jurisdiction over Medicinal Cannabis is ANVISA, which still has not regulated the matter.
Federal Police, Civil Police and Army also have jurisdiction over Medicinal Cannabis, once it is a forbidden substance in Brazil.
14. What is the regulatory framework for the authorization, pricing, and reimbursement of Medicinal Cannabis?
As mentioned above, Medicinal Cannabis is still forbidden in Brazil and so there is no regulatory framework about its authorization, pricing and reimbursement yet.
15. How is the production and import of Medicinal Cannabis regulated and by which agencies/authorities?
The procedure for manufacturing and importing Cannabis Products is set forth in Resolution RDC No. 327/19. However, as mentioned above, since Medicinal Cannabis is still forbidden in Brazil, in order to manufacture and commercialize Cannabis Products in national territory, the interested company shall import the pharmaceutical input into the forms of vegetable derivative, phytopharmaceutical, in bulk, or industrialized product.
16. What approval or notifications are necessary to produce or import Medicinal Cannabis?
Companies cannot import or produce Medicinal Cannabis in Brazil.
17. What is the regulatory framework for the marketing and distribution of Medicinal Cannabis?
There is no regulatory framework for the marketing and distribution of Medicinal Cannabis.
18. How can patients obtain Medicinal Cannabis?
Patients cannot obtain Medicinal Cannabis. However, according to Resolution No. 327/19, they must have prescription to obtain Cannabis Products, which is restricted to medical professionals legally registered before the Federal Medical Council.
Note that Cannabis Products may only be prescribed when there is no other therapeutic option available in the Brazilian market.
19. Who can prescribe Medicinal Cannabis?
The prescription of Medical Cannabis is forbidden. However, as mentioned in the previous question, Cannabis Products must be prescribed by medical professionals duly registered before the Federal Medical Council.
20. Is there a list of doctors authorized to prescribe Medicinal Cannabis?
N/A
21. What approvals or notifications are required to prescribe Medicinal Cannabis?
The prescription of Medical Cannabis is forbidden.
22. Where is Medicinal Cannabis available?
The prescription of Medical Cannabis is forbidden.
23. Is there a list of retailers authorized to sell Medicinal Cannabis?
N/A.
24. Are there proposals for reform or significant change to the regulation of Medicinal Cannabis?
No. At this moment, there are no proposals for reforming or significantly changing to the regulation of Medicinal Cannabis.
Opioid Drugs
25. Are Opioid Drugs authorized in your country?
Yes, Opioid Drugs are authorized since duly registered before ANVISA. Note that opioid substances are subject to special control (Ordinance No. 344/1998).
There are some opioid derivate substances that are forbidden in Brazil (List F1), for example, heroin.
26. What are the regulatory authorities with jurisdiction over Opioid Drugs?
The regulatory authority with jurisdiction over Opioid Drugs is ANVISA.
Federal Police, Civil Police and Army may also have jurisdiction over Opioid Drugs depending on the substance used on the drug, since it may be a controlled substance.
27. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Opioid Drugs?
No, there is no specific regulatory framework for the authorization, pricing, and reimbursement of Opioid Drugs.
The only resolution that may be used as parameter is Ordinance No. 344/1998, which provides for substances subject to special control.
28. Which are the Opioid drugs that have received market approval to date?
There are several drugs approved, on all of the four major categories: codeins, fentanyls, oxycodones and tramadols.
29. Who can prescribe Opioid Drugs?
The physician duly enrolled before the Medical Council.
30. Is there a list of doctors authorized to prescribe Opioid Drugs?
No.
31. What approvals or notifications are required to prescribe Opioid Drugs?
The physician must be duly registered before the Regional Medical Council.
32. Which organizations are authorized to sell/distribute Opioid Drugs available?
In order to commercialize Opioid Drugs the interested company must be licensed before ANVISA and local health authorities, and the product must be duly registered before the Agency.
33. Is there a list of retailers/distributors authorized to sell Opioid Drugs?
No.
34. Are there proposals for reform or significant change to the regulation of Opioid Drugs?
We are not aware about any change in the regulatory framework.
35. When are they likely to come into force?
N/A
Also from this Legal Handbook
5. Regulatory Reform: Brazil
A brief insight into upcoming regulatory reforms in Brazilian pharma. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. Are there proposals for reform or significant change to the healthcare system?
There are plenty of Law projects under discussion. Before National Congress, there is Bill No. 667/2021, which provides for the creation of Risk Sharing Agreement for the incorporation of new health technologies.
Furthermore, there are several relevant themes included on ANVISA’s Regulatory Agenda of 2021-2023, such as: (i) review of Resolution RDC No. 98/2016, which provides OTC drugs; (ii) review of GMP and Certificate of Good Practices for Distribution and Storage; (iii) review of the drug labelling requirements; (iv) review of Resolutions RDC 9/2015 and 10/2015, which provides for clinical trials with drugs and medical devices, respectively; (v) review of the requirements for proof of safety and efficacy of the synthetic drugs (Resolution RDC 200/2017); (vi) use of analysis performed by Equivalent Foreign Regulatory Authorities for regularization of products before ANVISA (GGMED); (vii) expanded Access to Medical Devices; (viii) reprocessing of medical devices; (ix) Good Practices for pharmacies; among others.
On March 30, 2021 ANVISA published the Resolution RDC No. 657/2022, which provides for the regularization of software as medical device (SaMD). It will come into force on July 1st, 2022.
2. When are they likely to come into force?
Unfortunately, it is not possible to predict if and when the Bill will come into force. Nonetheless, ANVISA’s regulatory agenda is planned to be concluded until 2023.
Also from this Legal Handbook
6. Patents and Trademarks: Brazil
An overview of the legal framework for patents and trademarks for pharmaceuticals in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. What are the basic requirements to obtain patent and trademark protection?
Industrial Property Rights are regulated in Brazil through Federal Law No. 9,279/1996 (“IP Law”).
Under its Article 8, the main requirements for the grant of a patent are novelty, inventive step and industrial application. The law provides other requirements such as sufficient description, and prohibitions, such as impossibility to add subject matter after the patent examination request is made, double protection, patents involving methods of treatment etc. Please note, however, that several other formal requirements are applicable, many of them being regulated by internal rules enacted by the Brazilian Patent and Trademark Office (BPTO).
As to trademarks, Brazil is a first-to-file country, meaning that the registration of a trademark is granted to the first person who files the application. Article 129 of the IP Law sets forth that the ownership of a trademark (and the exclusive rights of use thereof) is obtained through the issuance of a valid registration by the BPTO.
Article 124 of the IP Law states that the following are not registrable as trademarks:
- crests, armorial bearing, medals, flags, emblems, official public distinctions and monuments, be they national, foreign or international, as well as any respective designations, figures or imitations;
- an isolated letter, digit or date, except when sufficiently distinctive.
- expressions, figures, drawings or any other contrary to morals and good customs or which offend a person’s honor or image or are an affront to the liberty of conscience, beliefs, religious cults or to ideas and sentiments worthy of respect and veneration.
- signs of a generic, necessary, common, usual or simply descriptive character, when related to the product or service to be distinguished, or those commonly used to be designated a characteristic of the product or service with respect to its nature, nationality, weight, value, quality and moment of production or of giving service, except when present in a sufficiently distinctive manner.
- signs or expressions used only as a means of advertising.
- colors or their names, except when arranged or combined in an unusual and distinctive manner.
- reproductions or imitations of titles, bonds, coins and bank notes of the federal government, the States, the Federal District, the Territories, the Municipalities or of any country.
- technical terms used in the industry, science or art that is related to the product or service to be distinguished; and
- necessary, common or usual shapes of a product or of its packaging, or, furthermore, shapes that cannot be disassociated from a technical effect.
- designations or acronyms of a public entity or establishment, when registration is not requested by that public entity or establishment.
- reproductions or imitations of a characteristic or differentiating element of a title of an establishment or the name of an undertaking belonging to a third party, which are likely to cause confusion or association with such distinctive signs.
- geographic indications, imitations thereof likely to cause confusion or signs that might falsely suggest a geographic indication.
- signs that suggest a false indication with respect to origin, source, quality or utility of the product or service to which the mar is directed.
- reproductions or imitations of official seals, normally adopted for the guarantee of a standard of any type nature.
- reproductions or imitations of signs that have been registered as a collective or a certification mark by a third party, without prejudice to the provisions of article 154 [the collective and certification mark that have already been used and whose registrations are extinct cannot be registered in the name of a third party, before 5 years, counted from the expiration date].
- names, prizes or symbols of sporting, artistic, cultural, social, political economic or technical official or officially recognized events, as well as the imitations likely to cause confusion, except when authorized by the competent authority or entity prior to the event.
- personal names or signatures thereof, family or patronymic names and images of third parties, except with the consent of the owner, his heirs or his successors.
- literary, artistic or scientific works, as well as titles protected by copyright and likely to cause confusion or association, except with the consent of the author or owner.
- reproductions or imitations, in whole or in part, even with additions, of a mark registered by a third party, to distinguish or certify a product or service that is identical, similar or akin, and which are likely to cause confusion or association with the third party’s mark.
- duplications of marks of a single proprietor for the same product or service, except when, in the case of marks of the same nature, they are presented in a sufficiently distinctive manner.
- objects that are protected by industrial design registrations in the name of third parties; and
- signs that imitate or reproduce, wholly or in part, a mark of which the applicant could obviously not fail to have knowledge in view of his activity, and of which the proprietor is established or domiciled in the national territory or in a country with which Brazil maintains an agreement or guarantees reciprocity treatment, if the mark is intended to distinguish a product or service that is identical, similar or akin, and is likely to cause confusion or association with such third party mark.
Additionally, Articles 125 and 126 of the IP Law prohibit the registration of marks that reproduce or imitate famous and well-known marks, respectively.
2. What agencies or bodies regulate patents and trademarks?
Patent and trademarks applications are analysed and granted by the BPTO in the administrative sphere.
As to the judicial sphere, Brazilian courts are divided in two main jurisdictions, State and Federal courts. Generally, State courts deal with patent/trademark infringement cases, while validity issues are ruled by Federal courts, since the BPTO is a mandatory party in cases in which its administrative acts are being challenged.
3. What products, substances, and processes can be protected by patents or trademarks and what types cannot be protected?
Under the Brazilian IP Law, all products, substances and processes can be protected, except for those that incur on the prohibitions of Articles 10 and 18 of the IP Law, being worth mentioning: “operating or surgical techniques and therapeutic or diagnostic methods, for use on the human or animal body”; “natural living beings, in whole or in part, and biolog¬ical material, including the genome or germ plasma of any natural living being, when found in nature or isolated therefrom, and natural biological processes”; “that which is contrary to morals, good customs and public security, order and health”; “substances, matter, mixtures, elements or products of any kind, as well as the modification of their physical-chemical properties and the respective processes of obtaining or modifying them, when they result from the transformation of the atomic nucleus”.
According to Article 128 of the IP Law, applicants may only request the registration of a mark relating to the activity they effectively and licitly exercise directly or through entities that they control directly or indirectly. Further, the specification of products and services must not be too general, as this can lead to the BPTO to issue an office action for the applicant to explain it or restrict it. The Office can also (i) remove products/services if they are not deemed to be included in the claimed class; and (ii) rephrase the specification for accuracy.
4. How can patents and trademarks be revoked?
Patents may be revoked through an administrative invalidity request or court action, if one of the legal requirements for its validity are not present. Moreover, under Article 78 of the IP Law, patents may be revoked when the term of protection ends; on waiver by the patentee, without prejudice to the rights of third parties; on forfeiture; on non-payment of the annual fee; and on non-observance of the provisions of Article 217 (“a person domiciled abroad must maintain permanently a duly qualified attorney resident in the country, with powers to represent him administratively and judicially, including for receiving summons”).
Brazil also has specific rules on compulsory licences, which have been affected by the COVID-19. A Bill was approved to change the Patent Statute. It provides that in cases of national or international emergencies (and other scenarios) a compulsory licence may be granted ex officio if the patent holder or licensee does not meet the market’s needs. The new provisions also include a rule that for humanitarian reasons and if in accordance with a treaty to which Brazil is a party, compulsory licences may be granted for products that are destined to be exported to countries with insufficient or no capacity to manufacture the pharmaceutical product for the local population.
Revocation of a trademark registration may be pursued through:
- administrative invalidity request before the BPTO (Article 168 and following Articles of the IP Law). Such action can be filed within 180 days from the grant date and must include a statement of grounds. Supporting documents, if not attached to the action, may be submitted within 60 days from its filing date.
- non-use cancellation actions before the BPTO. Such action may only be filed after 5 years from the grant of the registration and must include a statement of grounds.
- court invalidity cases before Federal Courts. Such action can be filed within 5 years from grant of the registration. Having filed an administrative nullity action with the BPTO is not a condition for filing the court nullity action.
5. Are foreign patents and trademarks recognized and under what circumstances?
Foreign patents do not have a direct effect in the Brazilian territory, since patent rights are regulated by the territorial principle. Therefore, for producing effects in Brazil, the patent holder must file and prosecute a counter-part in the country, which will be analysed by the BPTO.
Foreign trademarks are recognized in Brazil if they are considered as well-known in their field of activity. Such marks enjoy special protection under Article 6 bis (1) of the Paris Convention and Article 126 of the IP Law, regardless of whether they have been previously filed or registered in Brazil. There are no certification proceedings for well known marks. The holder must oppose an application for a mark identical or similar to its well-known mark, or an administrative nullity action against a registration granted for a mark identical or similar to its well known mark, and submit evidence of its well known status (so that the applicant for the infringing mark could not have ignored it). If the well-known mark is not applied for or registered in Brazil, the holder must file an application within 60 days from the opposition/administrative nullity action date, and submit the filing receipt to the BPTO. The BPTO may reject, ex officio or at the outcome of an opposition, an application for a mark that wholly or partially reproduces or imitates a well-known mark.
6. Are there any non-patent/trademark barriers to competition to protect medicines or devices?
A regulatory barrier in Brazil is the data package exclusivity, although its application by Brazilian Courts is controversial. Such protection is provided for in Article 39.3 from the TRIPs, which was internalized in Brazil by Decree No. 1,355, dated of 12/31/1994, and is considered internal law, and in Article 195, XIV, of IPL, as follows:
“SECTION 7: PROTECTION OF UNDISCLOSED INFORMATION
Article 39
3. Members, when requiring, as a condition of approving the marketing of pharmaceutical or of agricultural chemical products which utilize new chemical entities, the submission of undisclosed test or other data, the origination of which involves a considerable effort, shall protect such data against unfair commercial use. In addition, Members shall protect such data against disclosure, except where necessary to protect the public, or unless steps are taken to ensure that the data are protected against unfair commercial use.”
Article 195 – A crime of unfair competition is committed by he who:
XIV – divulges, exploits or uses, without authorisation, the results of tests or other undisclosed data the elaboration of which involved considerable effort and which has been presented to government entities as a condition for approving the commercialization of products.”
Although the judicial recognition of data package exclusivity is still controversial in Brazil, especially due to the lack of a term of protection, there are judicial decisions applying the rule of Article 39.3 of TRIPs, in order to prevent the National Health Surveillance Agency (“ANVISA”) from granting registrations to copies, by “relying”, even if indirectly, from the dossier of the reference product.
Another regulatory barrier in Brazil is set forth in Article 5, caput, of Federal Law No. 6,630/76, which deals with sanitary surveillance in Brazil. Such Article establishes that medicines, drugs, pharmaceutical inputs and related products may not have names, designations, labels or packages leading to error. Paragraph 1 of the same article also states that the adoption of an identical or similar name for products having different compositions is forbidden, even if they are from the same manufacturer.
Finally, regulation issued by ANVISA may also apply for specific types of medicines or medical devices.
7. Are there restrictions on the types of medicines or devices that can be granted patent and trademark protection?
There is no specific restriction on types of medicines or devices that can be granted patent protection, being the general exceptions those of Articles 10 and 18 of the IP Law, as mentioned above. Likewise, there is no specific restrictions on types of medicines or devices that can be granted trademark protection, except for the legal obligation arising from Article 128 of the IP Law, as described above.
8. Must a patent or trademark license agreement with a foreign licensor be approved or accepted by any government or regulatory body?
According to Article 211 of Brazil’s IP Law, all technology transfer agreements and IP license agreements must be registered by the BPTO. This procedure is a legal requirement for (i) remittance of royalties abroad, (ii) tax deductibility of payments, and (iii) validity of the agreement against third parties. The following categories of contracts are defined by the BPTO and shall be registered within the Office:
- Exploitation of Patents (EP): contracts involving the licensing of granted patents or patent applications filed with the BPTO.
- Use of Marks (UM): contracts involving the licensing of registered trademarks or trademark applications filed with the BPTO.
- Supply of Technology (ST): contracts which main propose is the acquisition of knowledge not protected by industrial property rights filed or granted by the BPTO, for the production of industrial goods and services.
- Technical Assistance (SAT): contracts establishing conditions for obtaining techniques, planning methods and programming, as well as research, studies and projects for execution or specialized services.
- Franchise (FRA): contracts aimed at obtaining a temporary concession of rights involving the use of trademarks, technical assistance services, together or not, with any other kind of transfer of technology necessary required to execution of its object.
- Assignment of patent, design and trademark – involves the transfer of ownership and is subject to registration when it involves a payment and the holder is a foreign entity.
[1] Please note that Brazil’s Central Bank requires a copy of the agreement duly recorded at the BPTO (or a decision from the office stating that the contract does not need to be recorded, if it fits a few exceptions foreseen by the law) to allow payments to be remitted abroad for technology transfer agreement or IP licenses.
Also from this Legal Handbook
7. Product Liability: Brazil
The ins and outs of product liability in Brazilian pharma. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. What types of liability are recognized in your jurisdiction?
The scope of liability for suppliers and the standards for consumer protection in Brazil are, in some cases, more severe than the consumer rules applicable in other countries.
The Brazilian Consumer Defense Code – CDC establishes a strict liability regime, which means that under the CDC, the consumer will not have to prove the agent’s fault, but only the connection between the damage and the action of the agent. As a general rule, if the offense was caused by more than one responsible party, all the suppliers may be held jointly liable.
Violation to legal provisions of the consumer protection legislation may also subject the supplier to administrative and criminal penalties, in addition to the need to indemnify damages or harm caused to consumer (civil liability).
2. How do these types of liabilities apply to the manufacturers of medicines and devices?
The strict liability will be applied to the manufactures of medicine and devices for the redress of damages caused to consumers by defects from design, manufacture, construction, assembly, formula, handling, presentation or packaging of products, as well as for the improper or incomplete information about their use and risks.
In addition, non-compliance with the CDC, such as publishing misleading advertisement or not conducting the recall of hazardous products, may bring about administrative and criminal sanctions.
3. Does potential liability extend to the manufacturer only or could claims extend to corporate executives, employees, and representatives?
Article 12 of the CDC states that national or foreign manufacturers, producers, constructors, and importers are liable, regardless of the existence of culpability, for the redress of damages caused to consumers by defects from design, manufacture, construction, assembly, formula, handling, presentation, or packaging of products, as well as for the improper or incomplete information about their use and risks.
The seller shall equally be held responsible in the terms of the preceding article when:
- the manufacturer, constructor, producer or importer cannot be identified;
- the product is offered without a clear identification of its manufacturer, producer, constructor or importer;
- improper handling of perishable goods.
On the other hand, the personal responsibility of independent professionals shall be determined upon verification of the fault.
In addition, article 28 of the CDC states that the judge may disregard the legal identity of a company when, to consumers’ disadvantage, there is abuse of rights, excessive power, breach of the law, illicit act or fact, or violation of the bylaws or social contract. It shall be also exercised in the event of bankruptcy, insolvency, closing down, or inactivity of the body corporate resulting from mismanagement.
Also, the CDC considers certain practices as crimes against consumer relations, providing penalties such as imprisonment from three months to two years and/or payment of fines. Misleading and abusive advertisements are considered crimes against consumer relations. In the same way, not performing a recall campaign is understood by the CDC as a crime.
Federal Law No. 8,137/90, which defines crimes against the tax and economic systems, also provides for crimes against consumer relations, establishing penalties such as imprisonment from two to five years and/or fines. Some examples given by this law are: (i) selling or exposing for sale goods that are not in compliance with legal requirements or that do not fit their official classification and (ii) selling or storing product with improper conditions for consumption.
Please note that article 75 of the CDC states that those who anyway contribute to the crimes referred to in this Code shall incur in the applicable penalties according to the extent of their culpability. Likewise, the director, administrator, manager of the corporate body that promotes, allows or anyway approves the supply, offer, exhibition to sale or maintenance in store of the products or the rendering of services in the conditions hereby forbidden.
4. How can a liability claim be brought?
Federal and state district attorneys as well as Brazilian nongovernmental organizations (NGOs) registered with public record offices have standing orders to sue suppliers for damages caused to consumers individually and/or collectively, in view of non-compliance with the consumer protection legislation, in public civil actions regulated by Federal Laws No. 7,347/85 and No. 8,078/90.
Consumer protection agencies at the state and federal levels have concurrent jurisdiction to implement the consumer protection policy, as well as to sanction companies, in the administrative sphere, in case of non-compliance with consumer protection rules.
Generally, administrative penalties against suppliers include fines, product seizure, destruction of the product, cancellation of product registration with the competent authorities, prohibition to manufacture the product, suspension of product or service supply, temporary suspension of the activity, revocation of concession or permission to use, cancellation of license for the establishment or activity, total or partial closing down of the establishment, work or activity; administrative intervention and imposition of counter-advertising. Please note that such penalties may be applied cumulatively.
The penalty most commonly applied is the fine. Such penalty is graduated according to the seriousness of the infraction and the advantage obtained, and ranges from BRL 646.33 to BRL 9,691,976.50. Please note that this amount is updated on a quarterly basis.
Most of the public civil actions filed in Brazil were and are initiated by federal or state district attorneys’ offices. In these cases, district attorneys initiate civil inquiries prior to filing public civil actions, in order to evaluate whether the supplier has caused damage to consumers or not. In the civil inquiry procedure, companies usually have the opportunity to present the appropriate information regarding the investigation.
It is also possible to reach an agreement during the civil inquiry procedure. Such agreement, known as “Consent Decrees” normally establishes corrective practices to be adopted by the suppliers, which, in case of non-compliance will be subject to fines. Consent Decrees also usually have a clause that provides for the payment of an indemnification for the damage caused to the consumers, collectively.
As to individual civil liability lawsuits against suppliers, the CDC provides procedural tools to be used for the protection of consumers. In this scenario, the lawsuit may be filed in the consumers’ residence jurisdiction, once it facilitates the access to the judiciary.
Also, according to the CDC, consumers have as one of their basic right the facilitation for the defence of their rights, including the inversion of the burden of proof in their favour.
Please note that a criminal action could be promoted by the Attorney General Office as well, independent of the manifestation of the victim, in the crimes indicated in the article 61 to 74 of CDC and in the Federal Law No. 8,137/90.
Governmental entities and agencies specifically designed for the defence of the consumers interests and associations that have been legally constituted for at least one year whose institutional purposes include the defense of the interests of the consumers, may intervene as assistants to the Attorney General Office and are also empowered to propose a subsidiary penal action in case the charge is not brought forth within the legal term.
5. What defenses are available?
Article 12 of the Consumer Defense Code list three basic defenses that the manufactures could present:
- they did not introduce the product into the market;
- •he alleged defect does not exist; or
- the damage was caused solely by the consumer or third parties.
This list of defenses has been the subject of many legal discussions, including whether it is exhaustive or not. For example, another accepted defense is external force majeure, which is any event beyond the producer’s control that occurs after the product has been put on the market.
From the administrative perspective, depending on the circumstances and the sanctions applied against the suppliers, the annulment of the procedure can be requested. Relevant regulations establish formal and material elements that must be included in the infraction notices issued by the Agencies. The analysis of these rules is essential when constructing the claim of the annulment of an infraction notice or process.
Also from this Legal Handbook
8. Traditional Medicines & OTC Products: Brazil
A guide to the state-of-play regarding traditional medicines and over-the-counter (OTC) products in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. What are the regulatory requirements for traditional, herbal, complementary, or alternative medicines and devices?
Resolution RDC No. 26/2014 provides for traditional and herbal drugs requirements. These drugs are subject to registration, simplified registration (those included on the list of the Normative Instruction No. 2/2014 or on the Community herbal monographs with well-established use) or notification (those with active ingredient contained in the Herbal Medicines National Formulary of Brazilian Pharmacopoeia and Pharmacopoeias recognized by ANVISA), depending on its formulation. In order to submit the product to ANVISA’s analysis is necessary to present proof of safety use (non-clinical and clinical toxicology) and of therapeutic effectiveness (non-clinical and clinical pharmacology) of the drug.
2. Can these traditional, herbal, complementary, or alternative products be advertised directly to the public?
Yes, but only those that are classified as OTC.
3. What health, advertising, and marketing claims may be made for traditional, herbal, complementary, or alternative products?
Herbal and traditional drugs must comply with labelling and packaging limitations set forth in ANVISA Resolution No. 71/2009. Labels must only contain accurate, adequate, and safe information, among other specific information depending on the type of product. Note however that all claims included on the products’ label must be based on the studies and/or on the pharmacopoeia.
4. What are the regulatory requirements for over-the-counter (non-prescription) medications?
The regulatory requirements will depend on the category of the drug.
Those that offer low risk to the consumers (and are included on the list of ANVISA’s Normative Ruling No. 106/2021) are exempted from registration and only need to comply with the procedure of notification, which required the submission of a reduced number of documents capable of attesting to the quality and safety of the drug.
As abovementioned (on chapter Regulatory, pricing and reimbursement overview, question #6), there is also the simplified procedure for the registration, which applies not only for generics, but also for similar, specific, dynamized, phytotherapeutic and biological drugs that have the same production line, same manufacturer, same technical and clinical reports, the same composition of another drug already registered by the regular procedure before ANVISA, but with a different name, labelling packaging and registration holder. The analysis/review by ANVISA of the documents is faster (when compared with the regular procedure) since the documents were already analysed by ANVISA on a previous (regular) procedure.
Finally, there is also the OTCs drugs that are subject to the regular procedure (provided for in ANVISA’s Resolution No. 200/2017, which determined the minimum requirements for obtaining and renewing registration of drugs with synthetic and semi-synthetic active principles, classified as new, generic and similar), through this procedure the complete dossier will need to be submit.
5. Are there any limitations on locations or channels through which OTC products may be sold?
Only pharmacies and drugstores, with technical responsible present during all time of the operation, can dispense drugs. Note that remote means such as telephone and internet are also allowed, provided that the pharmacies and drugstores observe the restrictions set forth in the applicable rules.
6. What health, advertising, and marketing claims may be made for OTC products?
According to ANVISA’s Resolution RDC No. 96/2008 companies may only insert information that is scientifically evidenced. ANVISA also allows:
- the use of expressions such as “safe”, “effective” and “quality”, provided they are complemented by phrases that justify the veracity of the information, which must be extracted from studies published in scientific publications duly referenced;
- the use of expressions such as “absolute”, “excellent”, “maximum”, “optimum”, “perfect”, and “total” related to the efficacy and safety of the product, when faithfully reproduced from studies published in scientific publications and duly referenced;
- making reference to the number of countries where the product is marketed and/or manufactured, provided that the countries are identified in the advertisement.
7. Can OTC products be marketed or advertised directly to the public?
Yes. There are however some limitations concerning the advertisement.
8. What is the mechanism by which a prescription-only product can be converted to an OTC product?
According to Resolution RDC No. 98/2016, the registration holder must submit a requirement to ANVISA.
9. What are the requirements for the importation of either traditional medicines or OTC products?
The most important ones are (i) be duly enrolled before the health authorities for the activity of importation; and (ii) obtain prior consent from ANVISA (request under Resolution No. 81/2008).
Also from this Legal Handbook
9. Marketing, Manufacturing, Packaging & Labeling, Advertising: Brazil
Everything you need to know about the marketing, manufacturing, packaging & labeling and advertising of pharmaceuticals in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. What is the authorization process for the marketing of new drugs, biologics, medical devices, over-the-counter medications, and other medicinal products?
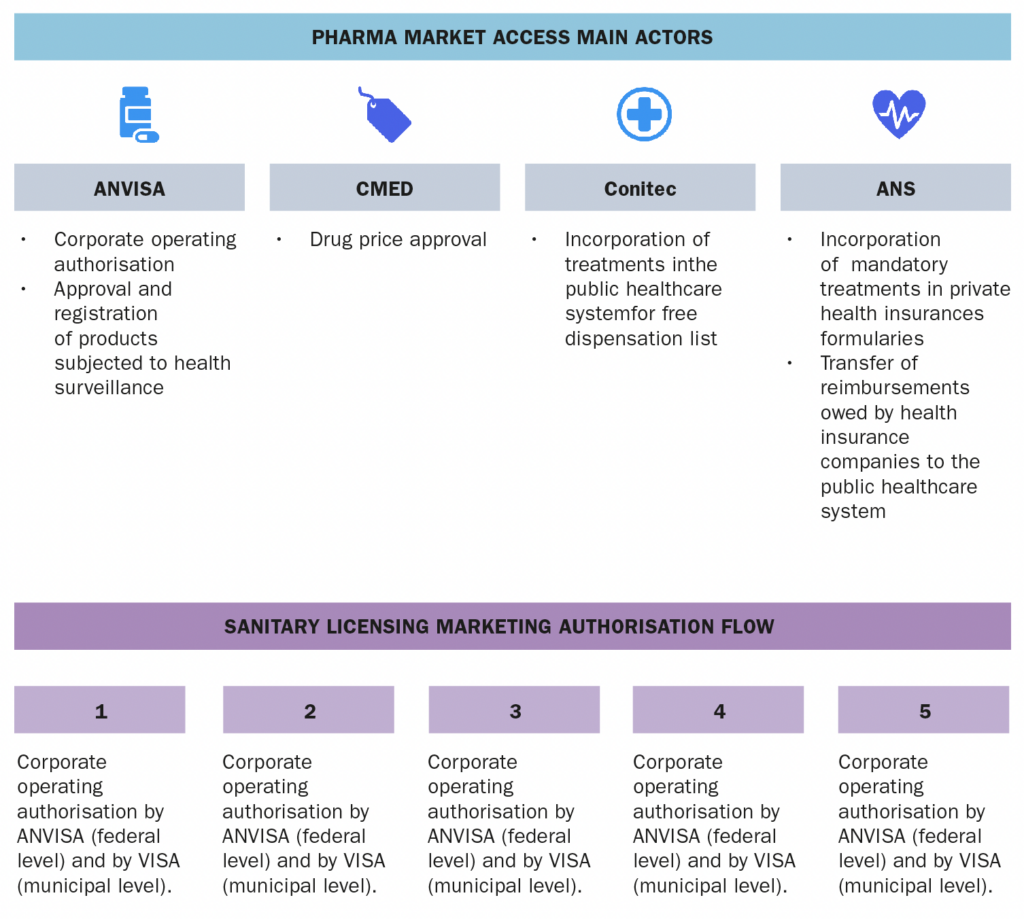
The company should be licensed before ANVISA (at the federal level) and before the local health authority (at the local level). The company must also be assisted by a technical responsible party. Finally, the product must be licensed by ANVISA and the company must obtain a GMP certificate (depending on the type of product). After being duly licensed by the health authorities, the company must apply for a product registration (the request must be attached to the product’s dossier, which contains detailed information about the overall development of the product, including efficacy and safety data, chemistry, manufacturing and control information and clinical trials). ANVISA will then review the information provided about the products and, if the analysis is positive, ANVISA will publish the issuance of the marketing authorisation in the official gazette.
2. What is the authorization process for the marketing of generic versions of these products?
The process is the same. Furthermore, as mentioned above (in chapter Regulatory, pricing and reimbursement overview, question #6), there is a simplified procedure for the registration of generic drugs, which is applicable only for drugs that have the same production line, manufacturer, technical and clinical reports, and composition as the drug used as a reference, that is already registered before ANVISA.
3. What are the typical fees for marketing approval?
The fees related to a product’s marketing authorisation will depend on the product and on the company’s corporate size (e.g. for new drugs the fee may vary from BRL 15,741.00 – approx. US$ 2,625.00 – up to BRL 157,416.00 – approx. US$ 26,236.00).
4. What is the period of authorization and the renewal process?
The company’s licenses from ANVISA do not expire.
A drug’s marketing authorisation is valid for 10 years and the renewal must be requested from six to twelve months before the product’s marketing authorisation expires. Marketing authorisation for medical devices is valid for 10 years.
The GMP certificate is valid for 2 years and the Certificate of Best Practices for Distribution and Storage of drugs is valid for 4 years.
5. What are the requirements, if any, for post-approval pharmacovigilance?
Pharmacovigilance is a duty of the marketing authorisation holder, which must comply with specific notification timings, depending on the type of issue, and must define measures and policies for identifying, evaluating, and monitoring the occurrence of adverse events and other issues related to commercialized drugs. The marketing authorisation holder is also responsible before ANVISA for recall measures.
6. Are foreign marketing authorizations recognized?
No.
7. Are parallel imports of medicines or devices allowed?
No. Only the marketing authorisation holder or a duly licensed legal entity properly authorized by the marketing authorisation holder may import a drug or a medical device.
8. What are the restrictions on marketing practices such as gifts, sponsorships, consultancy agreements, travel, and entertainment, or other incentives for healthcare organizations and individual medical practitioners?
Such practices are mainly ruled by ANVISA’s Resolution No. 96/2008 (in the event of drugs) and by industry association codes.
ANVISA Resolution RDC 96/08, article 5, provides that pharmaceutical companies cannot offer gifts, benefits or anything else of value to physicians who can prescribe medicines, whether or not the intent was quid pro quo. However, low-value gifts (pens, notebooks, etc.) are still authorized. Prescription pads cannot contain the company logo or promote a drug. Materials containing scientific information such as magazines and medical journals can be freely distributed.
The industry codes of conduct and the physicians’ professional councils impose additional restrictions on marketing activities and that anti-corruption laws also apply to public practice physicians.
Among industry codes, it is worth mentioning that the INTERFARMA (Pharmaceutical Research Association) code of conduct prohibits granting gifts offering gifts/benefits (i) of prescription drug; (ii) of a personal-use nature, including (but not limited to) electronic devices and/or tickets to concerts, theater, performances and sporting events; (iii) in cash and/or equivalent, including (but not limited to) credit cards, gift certificates and/or gift vouchers; (iv) of subscriptions to print and online magazines, even if they possess scientific contents; (v) used in the routine management of a medical office, including (but not limited to) pens, pencil-holders, and notepads.
The ABIMED (Brazilian Association of Industry of High Technology Medical and Hospital Equipment, Products and Suppliers) code of conduct provides that it is allowed to grant gifts that have an educational value, provided that such gifts (i) are related to the training of how to use certain product; (ii) are of low amount; and (iii) are related to the medical practice or provide benefits to a patient. Except for scientific books and anatomic models, the limit on gift amount is BRL 100 (approx. US$ 27.00).
Note that such Codes of Conduct are mandatory to the affiliated companies.
9. How is the manufacturing of medicines and devices regulated and by which agencies?
The manufacturing of drugs and devices is regulated by ANVISA, that authorizes the operation of the companies, issues resolutions related to good manufacturing practices, and performs inspections on company’s premises and products.
10. Are local manufacturing requirements compatible with Good Manufacturing Practices (GMPs) as defined by the U.S. Food & Drug Administration and/or the European Medicines Agency?
Yes, we consider that local requirements are compatible. ANVISA’s rules are influenced by North American and European rules.
11. What is the inspection regime for manufacturing facilities?
ANVISA and local health authorities have powers to perform inspections at any time in order to monitor company’s (i) regular licensing; and (ii) compliance with good manufacturing practices. Note that the authorities will always inspect companies’ premises when companies apply for the sanitary licenses or for the good manufacturing practices certification.
12. Are manufacturing facilities open for inspection by foreign inspectors or third-party inspectors as authorized by the FDA/EMA?
Yes.
13. What are the requirements for storage, packaging, and handling of medicines and devices and their constituent components?
The activities of storage, packing and handling of drugs are ruled by different federal and local rules. The type of drug may also influence the legal requirements. An evaluation on a case by case basis is necessary. As a rule, the storage must comply with the requirements established by product’s manufacturer. Regarding the label, Resolution RDC No. 71/2009, which rules labeling and packaging of drugs, provides that drugs’ packages must indicate: (i) commercial name of the drug; (ii) the generic name of each active principle; (iii) the concentration of each active principle; (iv) route of administration; (v) weight, volume or units; (vi) total quantity of dispensing accessories, where applicable; (vii) pharmaceutical form; (viii) use restriction by age group; (ix) qualitative and quantitative composition of each active principle; (x) conservation care; (xi) name, address and Taxpayer number of the marketing authorisation holder ; (xii) name and address of the manufacturing company, when the drug is imported; (xiii) name and address of the company responsible for the packaging; (xiv) name and number of enrolment before the Professional Council of marketing authorisation holder’s technical responsible; (xv) phone number of the Customer Service; and, (xvi) Number of the registration before ANVISA.
14. What information must be included in medicine and device labeling?
The requirements will vary depending on the type of product. However, in general, labels must contain information about importer’s and manufacturer’s corporate name and address, product’s commercial name, active ingredient, use instructions, warnings, batch, validity term, storage conditions, name of the technical responsible, number of registrations before ANVISA and customer service contact information.
15. What additional information may be included in labeling and packaging?
The requirements will depend on the type of the product.
16. What items may not be included in labeling and packaging?
The requirements will depend on the type of the product. Note, however, that labels must not contain false or misleading information.
17. What are the restrictions and requirements for the marketing and advertising of medicines and devices?
Some of the restrictions on the advertising of drugs are:
- The company which is promoting the advertisement must be duly licensed before the health authorities (Article 3º, §1º of Resolution RDC No. 96/2008).
- The drug which is being advertised must be duly registered before ANVISA (Article 3º of Resolution RDC No. 96/2008).
- The company must not promote an off-label use or an unregistered device (Article 3º, §2º of Resolution RDC No. 96/2008).
- Product’s registration numbers before ANVISA must be indicated in the advertisement materials (Article 22 of Resolution RDC No. 96/2008).
- All clinical and therapeutic claims in advertising material related to the drug must be based on substantiated scientific data having clinical and statistical significance (Article 7 of Resolution RDC No. 96/2008).
- Information inserted in advertisement material must be coherent with the information provided to ANVISA and with duly published and approved technical literature and scientific papers, as well as in accordance with regulation and legal requirements (Article 3º, §2º of Resolution RDC No. 96/2008).
- All advertising material must be truthful and non-misleading, coherent and consistent with the visual, artistic, and copy plan (Article 14 of Resolution RDC No. 96/2008);
- Under prescription drugs could be promoted only to physicians and pharmacists (“HCPs”) (Article 27 of Resolution RDC No. 96/2008).
- Adverting of OTCs must contain warning messages, advertisement of OTCs must contain warning messages (Article 22 of Resolution RDC No. 96/2008).
- Companies are not allowed to provide, offer, promise or distribute small gifts, benefits or advantages to prescribing or dispensing HCPs and to the general public. There are exceptions, such as institutional small gifts (which do not advertise drugs), scientific materials, such as technical books and educational materials for professional update (Article 5º of Resolution RDC No. 96/2008).
Advertising of drugs for the consumers are allowed only for OTC drugs and if the drug is registered before ANVISA and by a duly licensed company.
18. Where can medicines and devices be sold or delivered? Can medicines and devices be sold or delivered via post?
Only pharmacies and drugstores can commercialize drugs for the consumers and these companies can deliver the product by remote means, such as telephone and internet, provided that the restrictions defined in the legislation are met. Note however that the delivery should only be made by the pharmacies’ or drug stores’ currier and not by Brazilian Post, since the Brazilian Post has no pharmaceutical responsible to supervise the activity.
19. What are the restrictions and requirements for electronic marketing and advertising via email, by the Internet, social media, and other channels?
For drugs under prescription, the advertising must be made only to the prescribing or dispensing HCPs. It must also contain a liability statement should be presented informing about the legal access restriction. OTC drugs can be advertised through electronic means, provided that requirements are observed. Some of these requirements are:
- The company must not promote an off-label use or an unregistered device (Article 3º, §2º of Resolution RDC No. 96/2008).
- Product’s registration numbers before ANVISA must be indicated in the advertisement materials (Article 22 of Resolution RDC No. 96/2008).
- The clinical and therapeutic claims in advertising material related to the drug are based on substantiated scientific data having clinical and statistical significance (Article 7 of Resolution RDC No. 96/2008).
- Information inserted in advertisement material are coherent with the information provided to ANVISA and with duly published and approved technical literature and scientific papers, as well as in accordance with regulation and legal requirements (Article 3º, §2º of Resolution RDC No. 96/2008).
- All advertising material are truthful and non-misleading, coherent and consistent with the visual, artistic, and copy plan (Article 14 of Resolution RDC No. 96/2008).
- The advertisement includes the warning messages required (Article 22 of Resolution RDC No. 96/2008).
Note, however, that the current rules do not provide for advertising via email and social media.
20. May medicines and devices be advertised or sold directly to consumers?
No. Drugs and devices should only be commercialized to consumers by pharmacies and drugstores and advertisements are allowed only for OTC drugs.
21. How is compliance monitored?
At the federal level (by ANVISA) and at the local level (by the local health authorities). In both cases it could be through inspections, spontaneously initiated by such authorities or after anonymous tips from consumers or competitors.
22. What are the potential penalties for noncompliance?
Warning, fines (varying from BRL 2,000.00 – approx. USD 420.00; up to BRL 1,500,000.00 – approx. USD 320,000.00), seizure and disposal of the product, shut down of facilities, cancellation of sanitary licenses, prohibition of advertising, suspension of sale, imposition of rectifying message and/or the suspension of advertising.
Also from this Legal Handbook
10. Preclinical and Clinical Trial Requirements: Brazil
The requirements for conducting preclinical and clinical trials in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. Are clinical trials required to be conducted locally as a condition (stated or implicit) for marketing approval?
No.
2. How are clinical trials funded?
Clinical trials are funded by sponsors (an individual or legal entity, public or private, who finances the trial).
3. What are the requirements for preclinical and clinical trial protocols? Who must approve the protocols?
Clinical trials are subject to approval from a CEP and, in certain cases, by CONEP and ANVISA.
4. What are the requirements for consent by participants in clinical trials?
Participants must sign an Informed Consent Form, which must contain all necessary information, in clear and objective language, so the participant and/or his legal representative clearly understand the research in which he/she is willing to participate.
It must contain, for example: a) justification, objectives and procedures that will be used in the research detailing the methods applied, notifying the possibility of their inclusion into control or experimental groups, when applicable; b) explanation of potential side effects and risks resulting from participating in the research, besides the benefits expected from such participation and presenting measures and cautions to avoid and/or lower effects and adverse conditions that may cause damage, taking into account the characteristics and context of the research participant; c) clarification regarding monitoring and assistance that the research participants are entitled to, including the benefits and monitoring following the closure and/or interruption of the research; d) granting of total freedom to the research participant to refuse to participate or to remove his/her consent at any stage of the research, with no penalty; e) assurance of confidentiality and privacy of the participants during all phases of the research; f) clarification regarding reimbursement and how expenses incurred by the research participants will be covered; and g) clarification of indemnification guarantees for any damages resulting from the research.
5. May participants in clinical trials be compensated?
Participants may not be compensated, except those involved in clinical trials in phase 1 and in bioequivalence studies.
6. How are participants in clinical trials protected and indemnified against any harm that arises as a result of participation in the trial?
According to Resolution 466/2012, which governs the conducting of clinical trials (from an ethical perspective), the Informed Consent Form must not contain any provision (i) excluding the sponsor’s/investigator’s/institution’s responsibilities; and/or (ii) that leads to a participant’s waiver of their rights to seek indemnification for any damage.
The abovementioned rule does not accept the limitation of responsibility and/or indemnification. The sponsor and the institution are liable for any damages, predicted or not, caused to the patients and with the informed consent contain any disclaimer of liability, such disclaimer may be deemed unenforceable and the judiciary system may impose indemnification due to the damages caused.
Also, the same rule states that the research must guarantee that the benefits of the project are available to research participants, such as access to the proceedings, products or research agents. It may be concluded that study drugs and/or clinical trials must be provided to the research participants even after termination of the clinical trial.
It could be understood that the sponsor must provide and pay for the necessary medical proceedings during the course of the research. This position was confirmed by Brazilian judiciary system, strengthening the obligation of the sponsor to finance the medical proceedings involved in the research. In other words, it is understood that the participant must receive the benefits of the results achieved by the research that he or she was part of, and they should not be used only as a means for achieving the sponsor’s objectives.
According to Resolution CNS No. 563/2017, which regulates the subject’s right to post-study access to clinical research protocols intended for patients diagnosed with ultra-rare diseases, the sponsor must undertake responsibility and guarantee to every subject at the end of the study free access to the best prophylactic, diagnostic and therapeutic methods shown to be effective over five years after its registration before ANVISA. This period is calculated from the issuance of the product registration or, for drugs, from the definition of the price before the CMED.
Also from this Legal Handbook
11. Regulatory, Pricing and Reimbursement: Brazil
A brief legal overview of the situation regarding the regulation, pricing and reimbursement of pharmaceuticals in Brazil. Prepared in association with Trench, Rossi e Watanabe one of Brazil’s most prestigious law firms, this is an extract from The Pharma Legal Handbook: Brazil, which can be purchased for GBP 119, here.
1. What are the regulatory authorities with jurisdiction over drugs, biologicals, and medical devices in your country?
In Brazil, the authorities are the National Health Surveillance Agency (“ANVISA”), which is a federal entity linked to the Ministry of Health, and local health authorities in states and municipalities.
2. What is the regulatory framework for the authorization, pricing, and reimbursement of drugs, biologicals, and medical devices?
The main Statutes are the Federal Law 6,360/1976 and the Federal Decree No. 8,077/2013, which provide for the surveillance of drugs, medical devices and related products, and others are subject.
Federal Law No. 6,437/1977 is an important law; it sets forth the violations to federal health legislation, establishes their respective penalties, and makes other provisions.
Also, Federal Law No. 10,742/2003 created the Drugs Market Regulation Chamber (CMED) in order to regulate the prices of the drugs commercialized in the country.
Also, there are many regulations issued by ANVISA related to the products it inspects. See the list with the most important regulations below.
- Operating Authorization:
- Resolution RDC No. 16/2014: Operating Authorisation (“AFE”) authorizes a company to store, distribute, pack, export, import, manufacture, repack and transport.
- Operating License:
- Depends on the State/Municipality in which the operating facilities are established. In São Paulo, for example, the Ordinance CVS No. 1/2020, provides for the registration/licensing of facilities that develop activities subject to health legislation.
- Product registration:
- Resolution RDC No. 200/2017: provides for synthetic drugs, including generics and biosimilars.
- Resolution RDC No. 205/2017 provides for orphan drugs.
- Resolution RDC No. 55/2010 provides for biological drugs and biosimilars.
- Resolution RDC No. 238/2018 provides for dynamized drugs.
- Resolution RDC No. 26/2014 provides for phytotherapy drugs.
- Resolution RDC No. 73/2016: provides for post-registration changes and cancellation of drug registration (this resolution is applicable for synthetic and semisynthetic drugs, including generics and biosimilars).
- Resolution RDC No. 505/2021: defines the criteria necessary to develop and to request the marketing authorisation of high technology products based on human cells and genes, called “advanced therapy medicinal products”.
- Resolution RDC 465/2021: provides a registration and authorisation waiver authorisationfor emergency use of COVID-19 vaccines purchased by the Ministry of Health.
- Resolution RDC 595/2022: provides for self-tests for detecting the COVID-19 antigen.
- Best Practices Certificate:
- Resolution RDC No. 301/2019: provides for the requirements for the issuance of the Certificate of Best Manufacturing Practices for drugs. Note that drug manufacturing is subject to compliance to best practices. Import companies must request certification from the manufacturing plants located abroad.
- Resolution RDC No. 497/2021: provides for the procedure to obtain the Certificate of Best Manufacturing Practices and the Certificate of Best Practices for Distribution and Storage of drugs.
- Resolution RDC No. 16/2013: provides for the requirements for best manufacturing practices of medical devices and in-vitro diagnostic devices.
- Resolution RDC 508/2021: provides for the best practices for human cells for therapeutic use and clinical research, and makes other provisions.
- Resolution RDC 606/2022: provides exceptional temporary procedures to obtain the Certificate of Best Manufacturing Practices for active pharmaceutical ingredients and drugs and health products linked to Coronavirus;
- Technical Responsibility Certificate
- Federal Law No. 6.360/1976: provides that health companies must maintain duly qualified technical personnel, in quality and in quantity, to adequately cover the company’s needs.
- Pharmacovigilance
- Resolution RDC No. 406/2020: provides for pharmacovigilance norms for the holders of registrations for drugs for human use.
- Technovigilance
-
- Resolution RDC No. 67/2009: requires the implementation of a technovigilance system.
- Promotion and Marketing
- Resolution RDC No. 96/2008: establishes the general rule regarding advertising drugs. Note that there are several restrictions for advertising drugs (i.e. prescription drugs may only be advertised in scientific publications, intended for healthcare professionals).
- Resolution RDC No. 60/2009: provides for the distribution of product samples.
- Resolution RDC No. 71/2009: rules the labeling of drugs. However, rules applicable to each type of drug will provide for specific labeling requirements.
Note that this is not an exhaustive list of the regulations related to drugs and medical devices, but only a list of the main applicable rules.
3. What are the steps to obtain authorization to develop, test, and market a product?
Clinical trials are subject to approval from the Committee for Ethics in Research (CEP) and, in certain cases, from the National Committee for Ethics in Research (CONEP) and ANVISA.
The research protocol document must be submitted to CONEP, or CEP, and must describe the trial’s purpose and other details. It must also include information about the research participants and the researchers’ qualifications and all responsible parties. One of the documents to be filed with the research protocol is the Informed Consent Form.
After the protocol is submitted to the CEP/CONEP,the relevant authority will analyze the request and if the clinical trial complies with the applicable legislation, the authority will authorize the commencement of the research.
In order to market a product (drugs, biologicals, and medical devices), it will be necessary to (i) obtain a company license from ANVISA (Operating Authorisation – AFE) and from the local health authorities (Operating License – LF); (ii) maintain a Responsible Technical Party for the company; (iii) apply for Best Manufacturing Practices certification, depending on the product, and (iv) obtain the product’s registration/enrolment before the ANVISA.
4. What are the approximate fees for each authorization?
The fees vary depending on the type of authorisation, on the product and on the company’s corporate size.
5. For how long are marketing authorizations/registrations valid? How are marketing authorizations/registrations renewed?
In general, marketing authorisation for drugs and medical devices authorisationis valid for ten years. In certain cases applicable to drugs, the period can be of three or five years, pursuant Resolution RDC 317/2019.
The renewal of both types of marketing authorisations must be requested from six to twelve months before the marketing authorisation expiration date, as set forth in Resolution RDC 250/2004.
6.A. How does the authorization process differ between brand-name products and generic products?
As a rule, the authorisation process does not change, but the documents and studies to be presented to ANVISA differ. The registration of generics will require studies in order to prove that the product is stable and therapeutically equivalent to the reference drug, with pharmaceutical equivalence and bioequivalence studies, while the registration of a new drug should also be attached to its clinical trial studies (with the studies necessary to prove the quality, safety and efficacy of the product).
Note that there is also the simplified procedure for the registration of generic drugs. This procedure only applies to drugs that have the same production line, manufacturer, technical and clinical reports, and composition of the drug used as reference, which is already registered with the normal procedure before ANVISA. The differences between the reference drug and the generic one are their name, labeling and packaging. Through this procedure the analysis/review by ANVISA of the documents is faster (when compared with the normal procedure), since the documents were already analyzed by ANVISA during the previous (normal) procedure.
6.B. Are there differences for local manufacturers versus foreign-owned manufacturers?
No, the authorisation process is the same. However, certain timings may differ (i.e, timings for foreign and local inspection for -Best Manufacturing Practices certification are different) and some additional documents will be required for imported products.
Note, however, that only duly licensed Brazilian legal entities may be the marketing authorisation holder.
7. How are combination products (drug + drug, drug + biologic, drug + device, biologic + device, drug + biologic + device) regulated?
Our understanding is that ANVISA will analyse a product’s purpose. For example with a drug + device, a product’s final registration will be for a drug and, therefore, the final product must be considered as a drug for all other regulatory purposes. However, there is no specific rule regarding this matter.
8. How is compliance with regulation monitored and evaluated? Is the regulatory regime comparable with the U.S. Food and Drug Administration or the European Medicines Agency expectations and requirements?
ANVISA and the local health authorities may perform routine inspections on the premises of the companies that store, distribute, pack, export, import, manufacture, repack and transport drugs and/or medical devices. ANVISA is currently trying to focus its actions on actual monitoring and on-site inspections rather than the limited documentation analysis it used to perform.
We consider that the regime is comparable, mainly to the European agency standards, and, in that sense, ANVISA and the FDA are involved in arrangements to strengthen existing structures and develop new opportunities for cooperative engagement in regulatory and scientific matters and public health protection that are related to the products ANVISA and the FDA regulate.
9. What is the potential range of penalties for noncompliance?
A warning, a fine (varying from BRL 2,000.00 up to 1,500,000.00), the appropriation and disposal of the product, shut down of facilities, cancellation of health licenses, prohibition of advertising, suspension of sale, imposition of a rectifying message and/or suspension of advertising.
10. Is there a national healthcare system? If so, how is it administered and funded?
Yes. As provided in the Brazilian Federal Constitution of 1988, health is a right for all and a duty of the state. The public healthcare system was introduced by the Brazilian Federal Constitution of 1988 and is called the Unified Health System (SUS). The SUS provides for free and universal coverage and is funded through the social welfare budget of the federal government, the states, the Federal District and the municipalities, as well as from other sources such as fines, fees and donations.
The Federal Constitution of 1988 also allows for the complementary existence of the private sector in health assistance.
11. How does the government (or public) healthcare system function with private sector healthcare?
In the public sector, healthcare is delivered through programs and plans that are implemented at federal, state and municipal levels. Actions and services must be organized and executed on a regional basis. The SUS is a complex program and is organized according to the different federative levels; the federal government is primarily involved in the general planning of the system, organisation of health assistance centres in remote regions, and to providing financial grants to state, municipal and private health entities that operate in coordination with the SUS. The states are responsible for high and medium complexity health assistance centres, and the coordination of the efforts of the municipalities. The municipalities are responsible for primary care and low complexity health assistance institutions.
The private health system operates in parallel with the public healthcare system, with full capacity to provide health assistance to the population, whether through out-of-pocket payments or through health insurance companies.
12. Are prices of drugs and devices regulated and, if so, how?
The prices of drugs are regulated. The Drug Market Regulation Chamber (CMED) establishes the maximum price applied to a drug, depending on the party in the marketing chain. The Chamber also determines the discount that the public entity should apply in order to purchase a drug.
Prices of other categories of products, such as medical devices, diagnosis equipment are not regulated.
13. How are drugs and devices used by patients paid for? What roles do public and private payers play?
The SUS provides financial support to private health institutions by financial grants and reimbursement of medical procedures, devices and drugs upon the signature of an agreement between the private entity and the Ministry of Health. The reimbursement values are listed, along with the types of procedure and therapy covered. The private system is funded by out-of-pocket payments made by individuals (users) or by private health insurance. The coverage of health insurances is also defined according to a list of covered therapies and medical procedures issued by the Supplementary Health Agency.
14. Who dispenses drugs and devices to patients and how are those dispensers compensated?
Pharmacies, drug stores and hospitals. The compensation can be through reimbursement values or by out-of-pocket payments made by individuals.
15. What are the professional and legal responsibilities of those who dispense drugs and devices? What role do they play in providing patient care, information, and safety?
The entities that dispense drugs and devices must be assisted by a technical responsible party, who undertakes responsibility for the compliance with all sanitary legislation. Violations of duties may lead to the imposition of administrative penalties (i.e, for the legal entity), criminal sanctions (i.e, to the individual involved in the violation), civil measures (i.e, indemnification claims) and professional sanctions (i.e, applied by the professional council to the technical responsible party).
Also from this Legal Handbook
12. Updates on Drug Distribution Regulation in Brazil
For more than 20 years, Ordinance No. 802/1998 (“Ordinance No. 802/1998”), issued by the Brazilian Ministry of Health, used to regulate the distribution of pharmaceutical products in Brazil. Needless to say that such rule, although very extensive, became outdated and stopped reflecting the dynamics and needs of those involved in the drugs supply chain.
For this reason and for a period of more than two years, the update of Ordinance No. 802/1998 was discussed by the Brazilian National Health of Surveillance Agency (“ANVISA”) and the interested entities through a Public Consultation.
Finally, after such a long discussion period, ANVISA approved a new Resolution to regulate the subject (Resolution RDC No. 304/2019) for providing for the “Good Practices of Distribution, Storage and Transportation of Drugs”. The Resolution aims at ensuring greater flexibility to the supply chain, control and traceability conditions, considering the existent technology.
Although published in September 2019, Resolution RDC No. 304/2019 will enter into force only in March 2021, except in relation to one specific article, through which ANVISA expressly authorizes the commercialization of drugs between distributors, if both distributors ensure the traceability of the product. This provision is a desired novelty as, under Ordinance No. 802/1998, distributors were only authorized to source from the marketing authorization holder.
In March 2020, Resolution No. 304/2020 was amended by ANVISA’s Resolution No. 360/2020 for considering a large number of requests submitted by the industry.
Main changes brought by Resolutions Nos. 304/2019 and 360/2020
When compared to Ordinance No. 802/1998, the main changes arising from Resolutions RDC Nos. 304 and 360 are the following:
- ANVISA authorized the commercialization of drugs between distributors, provided that the traceability of the product is maintained. Under the previous rule, distributors could only purchase drugs from the marketing authorization holder
- All parties involved in the supply chain will be responsible for the quality and safety of the drugs, including for recalls actions. According to the previous Ordinance, it was the sole responsibility of the producer.
- Allows the reintegration of the recovered drugs into the company’s stock after cases of robbery, theft or any other type of misappropriation. However, the reintegration may only be implemented after performing a technical evaluation of the product to evidence that there was no damage and that the quality, safety and efficacy of the drug is guaranteed. Exceptions apply.
- The Resolution authorizes the outsourcing of activities, provided that the authorization agreement is approved by the quality management and qualification system of the contracting party. In addition, the contracting party must provide the contractor with the information necessary to perform the contracted operations correctly, in accordance with the registration of the drugs and any other legal requirements.
Thus, it is perceived that despite creating new responsibility for all those which integrate the supply chain, the new Resolutions seek to ensure greater flexibility to activities related to distribution, adjusting it to market’s current expectations, as Ordinance No. 802/1998 served the completely different moment and technology of 20 years ago.
The flexibility and extension of regulatory responsibilities, however, increase the need of having sophisticated distribution agreements, covering not merely commercial matters, but also compliance topics and the regulatory obligations applicable to, for example, pharmacovigilance, recalls, quality matters and responsibilities regarding third parties (i.e, other distributors).