The Pharma Legal Handbook: The Netherlands




Ready to tap into the EUR 4.7 billion Dutch pharma market? The Pharma Legal Handbook: The Netherlands is your key to unlocking this strategically positioned and flourishing market.The Netherlands isn’t just about a robust pharma market; it’s a hub of top-notch institutions, including the European Medicines Agency, world-class healthcare, and a thriving start-up culture. Add to that a highly educated workforce, tax incentives, and innovation grants, and you’ve got a recipe for success.
Prepared with Houthoff, a top-tier Dutch law firm.
October 2023
1. Regulatory, Pricing and Reimbursement: The Netherlands
1. What are the regulatory authorities with jurisdiction over drugs, biologicals, and medical devices in your country?
Ministry of Health, Welfare and Sport (Ministerie van Volksgezondheid, Welzijn en Sport)
The policy and regulation of drugs (‘medicinal products’), biologicals and medical devices falls under the jurisdiction of the Ministry of Health, Welfare and Sport. The Ministry has the primary responsibility for the Dutch healthcare system. It has delegated most of its executive and supervisory tasks to governmental bodies.
Medicines Evaluation Board (College ter Beoordeling van Geneesmiddelen) (“MEB“)
The MEB is the governmental body that is responsible for the assessment of medicinal products, the registration of medicinal products, the issuing of marketing authorizations and the monitoring of adverse reactions and risks (i.e. pharmacovigilance).
Farmatec
Farmatec is part of the Central Information Unit on Healthcare Professions (Centraal Informatiepunt Beroepen Gezondheidszorg, (CIBG)), which is an implementing organization of the Ministry of Health, Welfare and Sport. Farmatec’s responsibilities include, the granting of pharmaceutical licenses (i.e. wholesale and manufacturing permits), the registration of certain medical devices, the setting of maximum prices for medicinal products, the issuing of Certificates of Pharmaceutical Products (CPP) for the export of pharmaceutical products, and the granting of exemptions for the marketing of opioid drugs.
Health and Youth Care Inspectorate (Inspectie Gezondheid en Jeudzorg, (IGJ)) (“Inspectorate“)
The Inspectorate is generally responsible for supervising compliance with the regulatory framework. The Inspectorate enforces the regulatory requirements concerning the safety of medical devices as well as medicinal products. Its tasks include the supervision of the ‘Good Practices’ throughout the supply chain, pharmacovigilance, the monitoring of adherence to the registration and authorization requirements, product recalls, and the requirements regarding inducement, transparency and marketing. However, responsibility for supervising compliance with the regulatory framework within the armed forces lies with the Ministry of Defense. Supervision is carried out by a military-medical inspection body. The Ministry of Transport and Water Management has primary responsibility for the requirements concerning the provision of medicinal products on board commercial shipping vessels. The Maritime Division of the Human Environment and Transport Inspectorate (Inspectie Leefomgeving en Transport) is tasked with the supervision and enforcement of these requirements.
Other
The Foundation for the Code for Pharmaceutical Advertising (Stichting Code Geneesmiddelenreclame (CGR)) lays down rules of conduct for prescription drug advertising and cooperation between healthcare providers and pharmaceutical companies.
The Healthcare Transparency Register Foundation (Stichting Transparantieregister Zorg) is an implementing organization that ensures the disclosure of financial relationships between healthcare providers and industry in a central register, the Healthcare Transparency Register (Transparantieregister Zorg). The Healthcare Transparency Register includes information provided by companies, healthcare providers and healthcare institutions pursuant to the Code of Conduct for Medical Devices (Gedragscode Medische Hulpmiddelen) (see further Question 2 of this Chapter) and the Code of Conduct for Pharmaceutical Advertising (Gedragscode Geneesmiddelenreclame) (see further Chapter 3, Question 17).
2. What is the regulatory framework for the authorization, pricing, and reimbursement of drugs, biologicals, and medical devices?
Drugs (medicinal products)
The Medicines Act (Geneesmiddelenwet) contains the requirements concerning the production and marketing of medicinal products and also provides for the implementation in Dutch law of Directive (EU) 2001/83 on medicinal products for human use. Implementing measures follow from the Medicines Act Regulation (Regeling Geneesmiddelenwet) and the Medicines Act Decree (Besluit Geneesmiddelenwet). The Medicine Prices Act (Regeling maximumprijzen geneesmiddelen) sets maximum allowable prices for medicinal products in the Netherlands. The rules on the reimbursement of medicinal products are tied in with the national health insurance system.
Biologicals
Biologicals are included in the Medicines Act. Therefore, biologicals are not addressed separately in the remainder of this contribution unless they are treated differently.
Medical devices
Regulation (EU) 2017/745 on medical devices (“MDR“) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (“IVDR“) apply directly to medical devices produced, placed on the market and offered to patients in the Netherlands (including via e-commerce). These regulations have direct effect and do not require transposition into Dutch law. The Medical Devices Implementing Regulation (Regeling medische hulpmiddelen) sets language requirements and designates competent authorities for the implementation of the MDR and the IVDR. Additional national requirements and requirements relating to the application of these regulations are laid down in the Medical Devices Act (Wet medische hulpmiddelen). The implementing rules relating to this act concerning medical devices are laid down in the Medical Devices Decree (Besluit medische hulpmiddelen) and the implementing rules concerning in vitro diagnostics are laid down in the In Vitro Diagnostic Medical Devices Decree (Besluit in-vitro diagnostica).
Moreover, a private Code of Conduct for medical devices aims to elaborate, in addition to the applicable legislation, on a careful, transparent and responsible relationship between suppliers of medical devices and the parties involved in the decision-making about their purchase and/or application. A sizeable number of medical device suppliers adhere to this code.
Reimbursement
Most medicinal products and medicinal devices are reimbursed on the basis of either the Healthcare Insurance Act (Zorgverzekeringswet) or the Long-Term Care Act (Wet langdurige zorg) and the accompanying regulations, e.g. the Healthcare Insurance Regulation (Regeling Zorgverzekering) and the Long-Term Care Regulation (Regeling langdurige zorg) (see further Questions 10 to 13 of this Chapter).
3. What are the steps to obtain authorization to develop, test, and market a product?
Drugs (medicinal products)
Development and testing
Authorization for new clinical trials (drug research) must be obtained from the Member State in which the drug research takes place, via the procedure prescribed by the Clinical Trial Regulation (EU) 536/2014 (“CTR“). The CTR entered into force on 31 January 2022 and replaces the Clinical Trials Directive 2001/20/EC. The Netherlands had implemented this Directive in the Medical Research (Human Subjects) Act (Wet medisch-wetenschappelijk onderzoek met mensen, ”MRA”). With the entry into force of the CTR, Directive 2001/20/EC was repealed, as well as Article 5a of the MRA, which specifically concerned drug research. From 31 January 2022 onwards, a three-year transition period applies for drug studies approved under the Directive 2001/20/EC and the MRA to the CTR. As from 31 January 2023, the CTR applies to all new applications for drug research. Under the CTR, a single procedure for submission, assessment and conduct of drug research applies to all drug studies (national and multinational) conducted in the European Economic Area (EEA). Clinical trial applications must be submitted via the Clinical Trials Information System (“CTIS“), which is maintained by the European Medicines Agency (“EMA“). The authorization and oversight of clinical trials remains the responsibility of the Member States. The CTIS is maintained by the EMA and is the single entry point for submitting the clinical trial application in the EU. From 31 January 2025 onwards, all current drug studies that have been approved under the old framework must comply with the CTR requirements as well. The MRA requirements remain applicable to other forms of medical research that do not fall within the scope of the CTR (see further Chapter 2).
Manufacturing
Dutch law prohibits the preparation, import, stocking, offer for sale, delivery, export or otherwise bringing of products containing active pharmaceutical ingredients (“APIs“) into Dutch territory, or taking them out of Dutch territory, or the operation of a wholesale business in APIs, without prior registration (Article 38 Medicines Act). In practice, this registration obligation is implemented via a permit requirement. Permits are issued by Farmatec. Two different permits exist:
- A wholesale permit for the sourcing of APIs from an EEA country;
- A manufacturing permit for the preparation and procurement of medicinal products. In this regard, packaging and labeling are also considered preparatory acts for which a manufacturing permit is required. The sourcing of APIs from outside the EEA also requires a manufacturing permit. Finally, an additional permit is required for the preparation of medicinal products involving human tissue collected for human treatment, on the basis of the Safety and Quality of Body Materials Act (Wet veiligheid en kwaliteit lichaamsmateriaal).
Moreover, the ‘Good Practices’ must be adhered to throughout the supply chain for medicinal products that are destined for the EU market: the Good Laboratory Practices (“GLP“), Good Manufacturing Practices (“GMP“) and Good Distribution Practices (“GDP“) prescribe quality and safety standards and procedures for different stages of the supply chain.
Marketing authorization
Every medicinal product marketed in the Netherlands must have marketing authorization (“MA“) issued by a competent authority (Article 1(1)(III) Medicines Act). To obtain MA, an application must be submitted together with information and documents regarding the quality, safety and efficacy of the medicinal product. The competent authority for the EU, the EMA, based in Amsterdam, the Netherlands, or the competent authority for the Netherlands, the MEB, will carry out a benefit/risk assessment.
There are a number of exceptions from the requirement of MA, which are listed under (a) to (h) of Article 40 Medicines Act.
Procedure
In principle, applicants may choose one of the following procedures when submitting their application for MA, depending on the type of medicinal product and its intended marketing:
- the centralized procedurefor an MA valid in the EU;
- the decentralized procedurefor the marketing of the medicinal product on the markets of several EU member states at once by submitting an application to one Reference Member State (RMS);
- the national procedurefor an MA that is valid only in the Netherlands;
- the mutual recognition procedure(MRP) for an MA on the basis of recognition of the assessment by the RMS;
The application must be submitted by a natural person or legal entity established in the Netherlands or in another EU Member State. The EMA is responsible for the granting of marketing authorizations through the centralized procedure. The MEB is responsible for the granting of marketing authorizations through the decentralized procedure, the national procedure and the mutual recognition procedure.
The centralized procedure leads to one MA valid throughout the EU, which is its main advantage. Applicants must submit their application file to the EMA for assessment by the Committee for Medicinal Products for Human Use (CHMP). The centralized procedure is compulsory for human medicinal products containing a new active substance to treat, medicinal products derived from biotechnology processes, advanced-therapy medicinal products, orphan medicinal products, and veterinary medicinal products for use as growth or yield enhancers.
The decentralized procedure may be chosen if the applicant does not yet have an MA in any EU Member State or EEA State. It enables MA to be obtained for several EU/EEA states at once. The applicant may request one state to be the Reference Member State (RMS). An applicant opting for the Netherlands as RMS may use the MEB planning tool to select a time slot for the procedure.
The national procedure is available only if the centralized procedure is not required and if the applicant has not yet obtained an MA or submitted an MA application in another EU/EEA Member State. An MA obtained via the national procedure covers the Netherlands’ market only. The national procedure at the MEB may also serve as the first phase of an MRP if the Netherlands is going to act as the RMS in such procedure. In the MRP, the applicant applies for an MA in an EU/EEA Member State on the basis of the MA issued by the RMS. The RMS’s assessment forms the basis for the assessment in the other EU/EEA Member States.
If an application concerns a medicinal product for which the file is identical to that of a product that is already authorized in the Netherlands, the MEB can forego a full evaluation and issue a ‘duplex’ MA. In principle, this procedure can be followed for a medicinal product that fulfills both of the following: (i) it has an approved Risk Management Plan (RMP); and (ii) it received MA no more than 5 years ago or completed an MRP with the Netherlands as its RMS. The duplex procedure can be followed for medicinal products authorized more than five years ago if a specific set of supplementary conditions is fulfilled. The supplementary conditions aim to guarantee that the duplex and reference product remain identical.
There are several situations in which an applicant does not need to follow the standard application procedure. The main circumstances in which this is the case are listed below.
Abridged procedure:
- in certain cases, applicants are not obliged to submit the results of pre-clinical and clinical trials (Article 42(5) Medicines Act), for example as regards generic versions of medicinal products (see Question 6 of this Chapter).
- In the case of medicinal product shortages, an abridged procedure can also be applied under certain circumstances (Article 3.17a Medicines Act Regulation).
Other exceptions or additional requirements:
- In exceptional circumstances, the competent authority can grant a conditional marketing authorization.
- The MEB can allow the marketing of non-authorized medicinal products for compassionate use.
- For orphan medicinal products, market exclusivity applies after they receive an MA. For a period of ten years (which period can be extended by two years), no other MA may be considered or granted for the same therapeutic indication in respect of a similar medicinal product.
- Medicinal products prepared through magistral preparation by apothecaries are excluded from the authorization obligation.
- The MEB applies stricter requirements to the evaluation of medicinal products for children (pediatric use).
Medical devices
The MDR and the IVDR contain the conditions for placing medical devices on the market in any EU Member State.
Regarding the entity placing the medical device or in vitro diagnostic medical device on the market, the MDR and IVDR differentiate between manufacturers, importers, wholesalers and authorized representatives. If the manufacturer has its seat outside of the EU, an authorized representative must be designated that is ultimately responsible for communicating with the authorities and also liable in case of default.
The MDR divides medical devices into the following risk classes:
- Class I (low risk): products that are non-sterile or do not have a measuring function;
- Products of class Is: sterile medical devices;
- Products of class Im: medical devices with a measuring function;
- Devices of class Ir: reusable medicinal products;
- Class IIa (medium risk) and Class Iib (medium/high risk);
- Class III (high risk).
Development and testing
Clinical trials with medical devices must be notified to the Central Committee on Research Involving Human Subjects (CCMO), pursuant to the MRA (see further Chapter 2).
Marketing
Under European and Dutch law, there is no obligation to obtain an MA for medical devices and in vitro diagnostic devices. However, medical devices placed on the EU market must bear a CE marking, and manufacturers, or their authorized representative, are obliged to issue a declaration of conformity (“DoC“) before marketing a medical device. By issuing the DoC, the manufacturer acknowledges liability for the conformity of the medical device with the requirements of the MDR.
Conformity assessment
Class I medical devices are self-certified, which entails that the manufacturer itself may prepare a declaration of conformity with the MDR.
For medical devices that fall in Class IIa, IIb or III, a conformity assessment by an independent authority (“Notified Body“) is required for the CE marking of the product. If the Notified Body approves the product, the applicant will receive a CE certificate. The following three Notified Bodies have been appointed by the Dutch authorities: DEKRA Certification B.V., DARE!! Services B.V., and BSI Group The Netherlands B.V.
For certain high-risk devices, the Notified Body is required to consult an expert panel before issuing a CE certificate.
Registration
Before placing any medical device on the EU market, manufacturers located in the EU and authorized representatives of non-EU manufacturers may also be subject to registration requirements in EUDAMED, the online system developed by the European Commission for this purpose. If the relevant section of EUDAMED is not yet fully operational, manufacturers and authorized representatives located in the Netherlands must notify Farmatec of the device (via the online registration system NOTIS).
Netherlands-based manufacturers and authorized representatives of non-EU manufacturers are required to submit notifications to Farmatec in respect of medical devices and in vitro diagnostic medical devices belonging to specific risk classes:
- Class I medical devices must be registered in EUDAMED before placement on the market. If EUDAMED is not yet fully operational, the product must be registered in NOTIS. Existing notifications for Class I medical devices made under the MDR’s predecessor (Directive 93/42/EEC concerning medical devices) remain valid until 26 May 2024. Existing notifications must be entered into EUDAMED before that date to remain valid (or the NOTIS notification must be updated if EUDAMED is not yet fully operational).
- Custom-made medical devices within the meaning of Article 21 MDR must be registered in NOTIS (Article 2 Medicines Act Regulation). A list of matters that must be included in a notification is available on Farmatec’s website. The list includes address details, the means of identification of the device, a declaration that the device is exclusively intended for use by a specific patient or user, the specific characteristics of the product, and information on safety and performance.
- For medical devices that fall in Class IIa, IIb or III, notification is not required.
NOTIS cannot be accessed without a Level 2+ eRecognition token. ‘eRecognition’ is a digital tool for secure online communication between companies and Dutch government organizations. It is used for various affairs such as registration and permit applications. An eRecognition login token must be obtained from a private, recognized provider. A prerequisite for the use of eRecognition software is a registration number with the Dutch Chamber of Commerce. Therefore, the manufacturer or authorized representative must first ensure it is established in the Netherlands and entered in the commercial register of the Dutch Chamber of Commerce before it can initiate the required registration procedure.
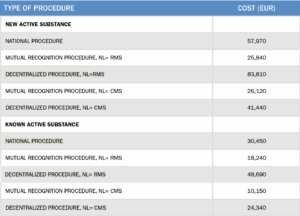
4. What are the approximate fees for each authorization?
Medicinal products
The fees for an MA application for a medicinal product are due from the moment of submission of the application and will not be refunded regardless of the outcome of the procedure. The fees charged by the MEB depend on the type of procedure and on the type of product. The MEB has published a full overview of the various fees on its website.
An overview of the application costs for the main categories is set out below (where RMS refers to Reference Member State and CMS refers to Concerned Member State):
The approximate fees payable for the examination of an application for medical device licence varies by the class of licence and the type of device, and range from $522 for Class II, $10,679 for Class III, to $25,955 for Class IV medical device applications.
Market authorizations for drugs are valid indefinitely, subject to their compliance with the Food and Drugs Act and its regulations.
SAP authorizations are valid only for maximum quantities of up to a six-month supply. Renewals can be ordered through the usual methods under the SAP.
Manufacturers of medical devices that are licenced for sale in Canada are required to inform Health Canada each year before November 1 that the information submitted with their licence application and any subsequent amendments have not changed. This is referred to as the licence renewal process. Manufacturers of licenced Class II, III and IV medical devices are charged an annual fee, payable at the time of licence renewal, for the right to sell their devices in Canada. The annual fee for the right to sell a licensed Class II, III or IV medical device in Canada is $394 as of April 1, 2022.
Persons holding an establishment licence (drug and device) also undergo a yearly review process, and must submit to the Minister, the information required in their original application each year. The licence holder will receive prior notification and failure to submit the review request will render the licence invalid.
The costs for amendments of an existing authorization are included in the annual fee, which amounts to EUR 1,900, or EUR 1,490 if the Netherlands serves as the RMS.
The MEB may suspend an MA if the annual fee is not paid in full in time, i.e. if the MA holder is in default under Dutch civil law (Article 6:81 Dutch Civil Code).
For the issuing of permits by Farmatec the following fees are due: for a manufacturing permit, an application fee of EUR 2,600 and an annual fee of EUR 3,200 (pro rata per month for the first year). For a wholesale permit, an application fee of EUR 1,700 and an annual fee of EUR 1,600.
Medical devices
If an assessment by a Notified Body is required, the manufacturer will bear the costs thereof. Notified bodies charge time-based or flat fees for the conformity assessment pursuant to the MDR, in accordance with standard lists made publicly available (Article 50 MDR and Article 46 IVDR).
Moreover, Article 111 MDR and Article 104 IVDR provide for the possibility of charging a fee for activities described in the regulations. On this basis, a fee is charged for the issuing of a certificate of free sale (outside of the EU) (Article 3 Medical Devices Implementing Regulation). Manufacturers or authorized representatives must pay a fee of EUR 62 per certificate and EUR 62 administration costs per invoice. Such certificate can be applied for via NOTIS.
A fee of EUR 200 applies for the notification to NOTIS of each individual product, regardless of whether the product is already registered in another EU Member State. No fee applies to product registration in EUDAMED. This fee does not include the cost of the eRecognition (see further Question 3 of this Chapter).
5. For how long are marketing authorizations/registrations valid? How are marketing authorizations/registrations renewed?
Medicinal products
In principle (unless a conditional marketing authorization as referred to in Article 45b Medicines Act is concerned), an MA for a medicinal product is valid for a period of five years from when it is first granted (Article 24(1) Directive 2001/83/EC). After five years, an authorization may be renewed at the request of the marketing authorization holder. The MEB will carry out a re-evaluation to determine whether the MA can be renewed, for which purpose the MA holder must submit a consolidated version of the file to the MEB, including changes made since the MA was granted and the review of suspected adverse reaction reports and periodic safety reports. The MA holder must do so at least nine months before the MA is due to expire. Renewal may be granted for an indefinite period, or for reasons of pharmacovigilance, for one more period of five years. The MA will be deemed to have lost its validity if the relevant medicinal product is not actually marketed for three consecutive years.
Medical devices
A DoC is valid indefinitely, but should be re-issued if significant changes to the medical device have been made. A CE certificate is valid for a maximum period of five years (Article 56 MDR).
6. How does the authorization process differ between brand-name products and generic products? Are there differences for local manufacturers versus foreign-owned manufacturers?
Authorization of generic products
For a generic product, the submission of a complete application file is not required; an applicant can apply for an abridged marketing procedure. The applicant is not required to provide results of pharmacological, toxicological and clinical tests if it is demonstrated that the product is a generic product (Article 42(5) Medicines Act). On this basis, the applicant may follow an abridged procedure with reference to the complete case file of the reference product. In evaluating the application, the MEB will verify whether the submitted product qualifies as a generic in comparison to the reference product.
In essence, the obligation to conduct pharmacological, toxicological and clinical tests that is part of the regular procedure is replaced by the obligation to demonstrate that the medicinal product is essentially equivalent to a medicinal product authorized not less than eight years prior to the date of the application by any EU Member State. That product is considered the reference medicinal product to the generic product. In the abridged procedure, the MEB will also evaluate the safety and effectiveness of the generic product.
The generic medicinal product may be marketed ten years after the grant of the MA for the reference medicinal product. This period can be extended by one year if the MA holder for the reference product has acquired a new pharmaceutical indication for which new pre-clinical and clinical studies have been conducted.
Authorization of brand-name products
For brand-name products, the procedure as set out under Question 3 of this Chapter applies.
Foreign-owned manufacturers
A manufacturer’s ownership is not decisive for the question of whether a medicinal product may be marketed in the Netherlands. Medicinal products for which an MA has been granted by the MEB, for which an MA issued by a different EU Member State has been recognized under the mutual recognition procedure, or for which MA has been granted by the Commission under the centralized procedure (see further Question 3 of this Chapter), may be placed on the Dutch market.
7. How are combination products (drug + drug, drug + biologic, drug + device, biologic + device, drug + biologic + device) regulated?
In principle, any substance or combination of substances intended to be administered, used or presented in any way as suitable for the treatment or medical diagnosis of a person is regarded as a medicinal product within the meaning of the Medicines Act (Article 1(1)(b) Medicines Act). This definition is in accordance with the definition of Directive (EU) 2001/83 on medicinal products for human use.
Products that combine a medicinal product or substance and a medical device are regulated under either the MDR or the Medicines Act, depending on the purpose of the product. If the medical device concerned incorporates, as an integral part, a substance that would in itself qualify as a medicinal product within the meaning of Directive (EU) 2001/83, but that primarily functions as and has the purpose of a medical device, the combination product is governed by the MDR. The Notified Body carrying out a conformity assessment of a medical device pursuant to the MDR or IVDR may request the MEB to advise on the quality, safety and beneficial properties of the integrated medicinal product (pursuant to Article 9(1)(h) Medicines Act). On the other hand, if the effect of the substance that qualifies as a medicinal product is most important, the combination product must be regarded as a medicinal product and the Medicines Act applies. The results of the assessment of the device part’s conformity with the relevant general safety and performance requirements pursuant to the MDR must be submitted in the MA procedure.
8. How is compliance with regulation monitored and evaluated? Is the regulatory regime comparable with the U.S. Food and Drug Administration or the European Medicines Agency expectations and requirements?
General supervision and enforcement
The Inspectorate, as the designated supervisory authority (see further Question 1), monitors the Dutch regulation of medicinal products and medical devices. To this end, it may carry out inspections and impose sanctions in cases of noncompliance. The Inspectorate also conducts periodic inspections of all drug manufacturers in the Netherlands, assessing whether the manufacturers comply with GMP and GDP (the Inspectorate usually combines GMP and GDP inspections). The Inspectorate may impose sanctions in cases of noncompliance. In principle, the Inspectorate publicly discloses enforcement documents, including inspection reports and sanction decisions (see further Question 9 of this Chapter).
Pharmacovigilance (medicinal products)
The requirements for pharmacovigilance are laid down in EU and national legislation and are further elaborated on in EMA’s good pharmacovigilance practices (GVP).
The MEB is the designated national competent authority for the coordination of pharmacovigilance in the Netherlands. It is responsible for the authorization and safety monitoring of medicinal products in the Netherlands (Article 76 Medicines Act).
With regard to its role in pharmacovigilance, the MEB is supported by the Netherlands Pharmacovigilance Centre ‘Lareb’, an independent foundation funded by the MEB and the Ministry of Health, Welfare and Sport. Patients and healthcare providers may report adverse drug reactions to Lareb via an online form. Lareb analyses reports of adverse drug reactions to identify risks associated with the use of medicinal products and reports its findings to the MEB for review.
The Inspectorate monitors compliance with the pharmacovigilance requirements by MA holders, and conducts inspections according to the national inspection program. It also carries out EU inspections in accordance with the EMA inspection program of central market authorization holders when the pharmacovigilance system master file (PSMF) is located in the Netherlands.
There are two types of inspections: routine inspections and unannounced inspections following a notification:
- routine inspections are scheduled in advance and usually focus on all elements of pharmacovigilance implementation; and
- inspections following a notification focus on specific elements.
For the national inspection program, the IGJ classifies MA holders according to their risk profile. This profile is generated through a pharmacovigilance questionnaire that MA holders fill out.
Regarding EU inspections, every EU MA holder should have a Pharmacovigilance System Master File (PSMF). This document contains all data and processes relevant to the marketing authorization holder’s pharmacovigilance system. This document must be registered in a European country. The Committee for Medicinal Products for Human Use (CHMP) has drawn up a European Inspection Plan for this purpose.
As of 1 February 2019, all reports resulting from the national inspection procedure are published by the IGJ. If an inspection shows noncompliance with the pharmacovigilance requirements by an MA holder, the Inspectorate will draw up a draft report and give the healthcare provider the opportunity to submit comments within a limited timeframe. The Inspectorate will amend the report if necessary and circulate the final version. The Inspectorate will publicly disclose the report via its website at least two weeks after the healthcare provider has received the final report. The Inspectorate informs the MEB and other Member States of cases of noncompliance.
Medical devices
Pursuant to the Medical Devices Act, Inspectorate officials are authorized to carry out inspections of compliance with the rules on medical devices and inspections of sanction violations.
Manufacturers and authorized representatives must report any serious incidents and corrective actions (Field Safety Corrective Actions) relating to medical devices or in vitro diagnostic medical devices to the Inspectorate within strict deadlines in accordance with the MDR. The Inspectorate publishes safety warnings that have been reported to it on its website. Reports will have to be made through EUDAMED once it becomes fully functional.
Inducement
The Inspectorate also enforces the rules on inducement that apply to medicinal products and medical devices on the basis of Article 94 Medicines Act and Article 6 Medical Devices Act (see further Chapter 3, Question 8). The Inspectorate monitors compliance with the inducement rule in accordance with the respective policy rules: the Policy Rules for Inducements under the Medicines Act 2018 (Beleidsregels gunstbetoon Geneesmiddelenwet 2018) and the Policy Rules for Inducements under the Medical Devices Act (Beleidsregels gunstbetoon Wet medische hulpmiddelen).
9. What is the potential range of penalties for noncompliance?
Administrative fines
An administrative fine may be imposed for each individual case of noncompliance with the rules on medicinal products and/or on medical devices, up to a maximum of EUR 870,000, or, if higher, a fine not exceeding 10% of the undertaking’s turnover for the preceding financial year. The method of determining the fine for each individual offense, the rules for the imposition of the fine and the maximum amounts are set out in the Annexes to the Ministry of Public Health Welfare and Sport Policy Rules on Administrative Fines 2019 (Beleidsregels bestuurlijke boete Ministerie Volksgezondheid Welzijn en Sport 2019). The level of a fine imposed under these policy rules is determined on the basis of a number of factors: the gravity of the offense, the risk class of the product (in as far as medical devices are concerned), the seriousness of the offense, the presence of mitigating circumstances, the culpability of the offender, the size of the company and the risk of recidivism.
Periodic penalty payments
Depending on the specific offense, a penalty payment may be imposed in addition to an administrative fine. The annexes to the policy rules mentioned above specify for which offenses the option of imposing a penalty payment is available.
Public disclosure
The Inspectorate is under an obligation to publicly disclose reports and other information concerning its enforcement of healthcare regulations, such as decisions of Dutch administrative law (under Article 5:2 of the General Administrative Act (Algemene wet bestuursrecht)) to impose a sanction, or to conduct enhanced surveillance of a MA holder. This obligation is subject to a limited number of exceptions, such as decisions to impose criminal sanctions. If a healthcare provider wishes to object to disclosure it may apply to the interim relief judge for a preliminary injunction within two weeks of receiving the final report. The compulsory public disclosure follows from Article 44 to 44e of the Health Act (Gezondheidswet) and applies since 1 February 2019. As of that date, the Inspectorate publishes the information it is legally required to disclose. No prior balancing of interests takes place.
Inducement
For infringement of the inducement rules, the inspection usually imposes an administrative fine. This fine can be imposed on both the supplier of the medical device and the medical professional or institution that accepts the inducement. The amount of the fine is calculated in accordance with the policy rules mentioned above. The standard amount for a fine for infringement of the prohibition of inducement is EUR 150,000.
Criminal sanctions
If an infringement is also classified as a criminal offense, the Inspectorate will inform the public prosecutor of the case of noncompliance (Article 6 of the Ministry of Public Health Welfare and Sport Policy Rules on Administrative Fines 2019).
10. Is there a national healthcare system? If so, how is it administered and funded?
Introduction
The Netherlands has a private healthcare system that is heavily regulated and monitored by the Ministry of Health, Welfare and Sport and public authorities including the Dutch Healthcare Authority (Nederlandse Zorgautoriteit (“NZa“), the National Health Care Institute (Zorginstituut Nederland) and the Inspectorate. Healthcare is provided via general practitioners, hospitals and other healthcare providers, who have to comply with a variety of rules and regulations on e.g. pricing, quality and certain medical standards.
In the Netherlands healthcare system, a distinction is made between cure and (long-term) care. The former is laid down in the Healthcare Insurance Act and is organized by (privatized) health insurers and financed on the basis of premiums and deductibles. The latter has its roots in the Long-Term Care Act; it is organized by a subsidiary of health insurers and financed by the government with designated funds received through taxation and income-dependent contributions. Some medical devices, e.g. wheelchairs and house adaptations, are financed by municipalities, based on the Social Support Act (Wet maatschappelijke ondersteuning). Depending on the individual situation, medicinal products and medical devices are reimbursed under one of these systems.
Basic health insurance
Dutch residents and employees have to take out basic healthcare insurance in accordance with the Healthcare Insurance Act. This type of healthcare makes up approximately 55% of the total healthcare costs in the Netherlands. Health insurers cannot refuse to provide basic insurance for any individual and there are certain restrictions in relation to the premiums charged for the insurance.
The Healthcare Insurance Act determines which care is eligible for reimbursement. Healthcare is mostly offered by contracted healthcare providers in kind. If insured individuals receive non-contracted care, insurers are not obliged to fully reimburse the costs. However, it is also possible to take out a more expensive insurance policy based on free choice of healthcare providers in which case non-contracted care is fully reimbursed as well. Academic hospitals receive additional contributions from the government, mainly for their academic activities. Specifically concerning medicinal products, only medicinal products included in the Drug Reimbursement System (Geneesmiddelenvergoedingssysteem, GVS) are covered (see further Question 13 of this Chapter).
Insurers conclude mostly annual contracts with healthcare providers to ensure their insured population has access to the basic care under the mandatory health insurance. Insurers receive monthly premiums and, depending on the health characteristics of the insured population, additional compensation from a special fund composed of designated taxation and income-dependent contributions. As of 2023, there are approximately 10 players in the Netherlands’ healthcare market, with a much larger number of insurance brands offering basic healthcare insurance.
Long-term care
Those insured are automatically covered for long-term care under the Long-Term Care Act. Long-term care makes up almost 33% of the total cost for healthcare. The coverage is paid fully by the government with money raised through designated taxation and income-dependent contributions. Subsidiaries of health insurers are responsible for organizing the long-term care and provide either the care or a payment for care contracted directly by the insured (via the ‘personal care budget’ (persoonsgebonden budget)). Whilst the long-term subsidiary has a duty of care towards its own insureds, for reasons of efficiency the long-term care is organized by one of the subsidiaries per region care administration offices (zorgkantoren). Both the subsidiaries and the care administration offices are fully reimbursed for the costs they make to organize long-term care.
11. How does the government (or public) healthcare system function with private sector healthcare?
Insurance companies offering basic health insurance usually offer various healthcare products that are complementary to the mandatory basic healthcare insurance. Such additional healthcare insurance is not governed by the Healthcare Insurance Act. As such, complementary healthcare insurance is not subject to an acceptance obligation. The health insurer determines the content, conditions and reimbursements of the supplementary insurance. Healthcare providers do have to comply with certain general rules and regulations, for example as regards quality.
12. Are prices of drugs and devices regulated and, if so, how?
Central government does not set fixed prices for medicinal products and devices. However, the Ministry of Health, Welfare and Sport does set maximum allowable prices for medicinal products twice a year by ministerial regulation in accordance with the Medicine Prices Act (Wet Geneesmiddelenprijzen). The Medicine Prices Act divides comparable medicinal products (intramural as well as extramural) in product groups based on API, strength and pharmaceutical form. The maximum allowable price for these product groups is then set on the basis of a comparison with the prices in four reference countries: Belgium, France, Norway and the United Kingdom. Germany was replaced by Norway as reference country for setting maximum prices by an amendment of the Act in April 2020, the primary reason being that prices in Germany were above the average for the Netherlands (and of the European Union as a whole) whereas the prices in Norway were significantly lower than in the Netherlands. At the same time, the social security system and level of healthcare were regarded as similar to the Netherlands. This amendment thus led to a decline in maximum prices in the subsequent evaluations of the maximum price.
The maximum price set for each registered medicinal product (brand name or generic), per quantity and pharmaceutical form, is included in the Annex to the Medicine Prices Act. The sale of a medicinal product at a price higher than the set maximum price, to end-users as well as to authorized suppliers of medicinal products to end users, is prohibited (Article 4 Medicine Prices Act). Sellers are under a legal obligation (Article 5 Medicine Prices Act) to keep records showing that they act in the conformity with the maximum price. Within these limits, the price may differ per health insurer, since wholesale margins are not fixed but rather are based on individual agreements resulting from negotiations. Health insurers negotiate vis-à-vis the manufacturer or wholesale distributor of the medicinal product on the one hand, and the pharmacists that dispense medicinal products to patients on the other.
13. How are drugs and devices used by patients paid for? What roles do public and private payers play?
Medicinal products are reimbursed or paid for directly by the health insurer (or zorgkantoor in the case of long-term care), unless the drug is not covered by the patient’s healthcare insurance or if the care falls under the compulsory deductible of the patient’s healthcare insurance, in which case the patient must pay for the drug themselves.
Moreover, Dutch health insurers will only reimburse a registered drug if it is included in the Drug Reimbursement System. The Ministry of Health, Welfare and Sport, and the National Health Care Institute decide together which medicinal products fall within the basic healthcare insurance coverage and whether they are either fully or partially reimbursable under the Drug Reimbursement System. The medicinal products included in the Drug Reimbursement System are divided in two lists which are included in Annex 1A and 1B of the Healthcare Insurance Regulation. Medicinal products which are interchangeable with each other are clustered and placed in Annex 1A. Medicinal products included in Annex 1A have a reimbursement limit. If the price exceeds this limit, the insured will have to pay for the additional sum. Medicinal products which are not interchangeable are placed in Annex 1B. No reimbursement limit applies to medicinal products included in this annex. Annex 2 of this regulation contains medicinal products which are only reimbursed under special conditions, such as for certain indications or patient groups. Over-the-Counter (OTC) products are not reimbursable.
Health insurers also generally only reimburse the cheapest version of a medicinal product containing the same active ingredient as the branded product. Health insurers are obliged to cover (at least) one variant per active ingredient. The preferential policy of the health insurer states for each active ingredient which variant is covered by the healthcare insurance. A different (more costly) variant will only be reimbursed by the health insurer if a healthcare practitioner has issued a statement that the patient requires a specific different variant for medical reasons.
The introduction of new medicinal products in the Netherlands takes a relatively long time as a price agreement has to be negotiated with the Minister of Health, Welfare and Sport before a medicinal product can be included in the Drug Reimbursement System. The supply of new medicinal products before such agreement is concluded is not reimbursed. However, pharmaceutical companies sometimes choose to make medicinal products available to patients (without receiving reimbursement) in anticipation of inclusion in the Drug Reimbursement System and pending an agreement on price and reimbursement.
14. Who dispenses drugs and devices to patients and how are those dispensers compensated?
Pharmacists dispense medicinal products on the basis of a prescription and must abide by legal rules and professional standards in doing so (see further Question 15 of this Chapter). Pharmacists may not pay more than the set maximum prices. When buying in medicinal products, pharmacists generally negotiate with the health insurers on the prices of medicinal products, which negotiations may lead to the conclusion of a contract between pharmacist and health insurer. If a medicinal product is included in the contract between health insurer and pharmacist, the medicinal product is paid for by directly by the health insurer. Otherwise, a patient must pay themselves and may subsequently submit the bill to the health insurer for full or partial reimbursement. In addition to the costs of the medicinal product, pharmacists may charge patients for services offered, which may include control of the prescribed strength and dosage, control of interaction with other medicinal products, delivery, and explanation of how to administer the medicinal product.
15. What are the professional and legal responsibilities of those who dispense drugs and devices? What role do they play in providing patient care, information, and safety?
To protect safety and patient care, the prescription of medicinal products is reserved to a limited group of healthcare professionals. This group includes medical practitioners, dentists and to a limited extent other healthcare professionals, on the basis of Article 36(14) Individual Healthcare Professions Act (Wet Beroepen in de Individuele Gezondheidszorg) (“BIG Act“). Healthcare professions based in the Netherlands, including practitioners and pharmacists, must register in the Register for Individual Healthcare Professions (Beroepen in de Individuele Gezondheidszorg register) and are subject to disciplinary rules (Article 3 and 4(2) BIG Act). Pharmacists must comply with many specific professional guidelines that are set out by the Dutch Royal Pharmacists Association (“KNMP“), such as the KNMP Guidelines on the Legal Framework for Professional Standards for Pharmaceutical Care. In particular, pharmacists must also abide by the ‘principle of good care’ as laid down in Article 40 BIG Act. The principle of good care is an open, ethical norm which is further elaborated upon in the KNMP Guidelines. These guidelines are used by the relevant authorities to check compliance with the norm. In effect, the principle of good care means that a pharmacist must, in general, personally dispense a medicinal product directly to the patient. Prescriptions from healthcare professionals established in other Member States must in principle be recognized by Dutch pharmacists on the basis of the Dutch implementation of Directive 2011/24/EU on cross-border healthcare (i.e. Article 6.14 Medicines Act Regulation), provided that the ‘principle of good care’ does not oppose such recognition.
Cooperation between practitioners and pharmacists is in principle prohibited in the Netherlands to the extent that such cooperation is not in the interest of the ‘good provision of medicinal product’ (Article 11 Medicines Act Decree). The Dutch Supreme Court has ruled in that regard that it is not in the interest of the good provision of medicinal product for general practitioners to set up a pharmacy if that leads to a conflict of interest. In the case in question, 75% of the general practitioners in a town were shareholders of the local pharmacy, and thus had a considerable interest in the turnover and profit of the local pharmacy.
Foreign prescriptions must be accepted by Dutch pharmacies if they contain the information as mentioned by the Annex of the Commission Implementing Directive 2012/52/EU (Article 6.14(2) Medicines Act Regulation). Moreover, prescriptions from healthcare professionals established in other EU Member States must in principle be recognized by Dutch pharmacists on the basis of the Dutch implementation of the Cross-Border Healthcare Directive (i.e. Article 6.14 Medicines Act Regulation), provided that the ‘principle of good care’ does not oppose such recognition. Online prescriptions are prohibited if the prescriber has never personally met the patient (Article 67 Medicines Act).
Also from this Legal Handbook
2. Preclinical and Clinical Trial Requirements: The Netherlands
1. Are clinical trials required to be conducted locally as a condition (stated or implicit) for marketing approval?
No, clinical trials do not have to be conducted locally as a condition for marketing approval. This is not one of the prerequisite conditions for approval of the clinical trial protocol, which conditions are listed in Article 3 MRA. Clinical trials may be carried out at different locations. The MRA only requires the clinical trial protocol to show that the clinical trial is carried out in appropriate institution(s).
2. How are clinical trials funded?
The entity responsible for the funding of the clinical trial, referred to as the sponsor, can be another entity than the investigator itself. Clinical trials may be funded privately (e.g. by a biopharmaceutical company), publicly (e.g. by a hospital or university) or by a combination of both. The sponsor plays a central role in the management of the clinical trial, which includes the application for authorization under the CTR (Article 5 CTR).
The CTR defines the sponsor as the individual, company, institution or organization responsible for the initiation, for the management and for setting up the financing of the clinical trial.
The MRA defines the sponsor as a person, company, institution or organization which provides for the research, which entails the assumption of the responsibility for the initiation, organization and/or funding of the research (Article 1(1)(f) MRA). The investigator remains responsible for the conduct of the clinical research at the clinical trial site.
3. What are the requirements for pre-clinical and clinical trial protocols? Who must approve the protocols?
CTR
For the Netherlands, the Central Committee on Research Involving Human Subjects (“CCMO“) is the entity responsible for the review of preclinical and clinical trial protocols. The review is carried out by either the CCMO itself or by a Medical Research Ethics Committee (“MREC“) that has been accredited by the CCMO.
Research protocols that concern a new clinical trial within the meaning of the CTR (Article 2(2)) must be included in the application file which must be submitted via CTIS (see further Chapter 1, Question 3). Review takes place in accordance with the CTR. The Clinical trial protocols falling within the scope of the CTR must contain at least the information included in Annex I part D of the CTR.
MRA
Research protocols for research to which the CTR does not apply, but which fall within the scope of the MRA, must meet the requirements of and contain the information stipulated by Article 3 MRA. Research protocols are reviewed in advance by the reviewing committee (Article 2 MRA). The research may not be carried out unless the reviewing committee has issued a positive opinion.
The MRA applies if the following two conditions are met:
- the protocol concerns medical scientific research, excluding drug research falling under the CTR; and
- the research involves the imposition of actions or rules of conduct on persons which affect those persons’ physical and/or psychological integrity.
4. What are the requirements for consent by participants in clinical trials?
CTR
Informed consent is one of the core prerequisites for the enrolment of any person in a clinical trial under the CTR. A person, or where the person is not able to give informed consent, their legally designated representative, must be informed in accordance with the requirements of Article 29(2) to 29(6) CTR. This entails that:
- the person, or their legally designated representative, must be given information enabling the understanding of:
- the nature, objectives, benefits, implications, risks and inconveniences of the clinical trial;
- rights and guarantees regarding their protection, in particular the right to refuse to participate and the right to withdraw from the clinical trial at any time;
- the conditions under which the clinical trial is to be conducted, including the expected duration of participation; and
- the possible treatment alternatives (including follow-up measures if participation is discontinued);
- the information must be understandable to a layperson;
- the information must be provided in a prior interview and must also be prepared in writing, which is available to the subject (or their legally designated representative). During the interview, it must be verified that the subject has understood the information;
- information must be included on indemnification of any harm which may result from the clinical trial (see further Question 6 of this Chapter); and
- the subject must be informed that a summary of the results of the clinical trial and a summary presented in terms understandable to a layperson will be made available in CTIS.
MRA
Likewise, medical research which falls within the scope of the MRA is prohibited without prior written consent, which is to be provided in accordance with Article 6 MRA. Consent must be given in the following manner:
- if the participant has not yet reached the age of twelve, written consent of the legal parent or guardian is required.
- for participants aged between twelve and sixteen, both the participant and the legal parent or guardian must give written consent.
- participants who have reached the age of sixteen must give their consent in writing (this can be done electronically).
- if the participant is twelve years and older and is not capable of a reasonable appreciation of their interests, the legal parent or guardian must give consent.
- in the case of a participant aged sixteen who is not capable of a reasonable appreciation of their interests, the participant’s legal representative must give consent. In the absence of a legal representative, the spouse, registered partner or other life companion of this individual may also give consent.
Before consent is sought, the person must be informed of the following:
- the purpose, nature and duration of the research;
- the health risks associated with participation in the research;
- the risks interim termination would entail for the health of the subject of the research;
- the drawbacks which the research might entail for the person; and
- the manner in which consent may be given.
The information must be provided in such a way that it is reasonably certain that the participant has understood its contents and must be provided in writing at the request of the participant and/or in an interview prior to consent.
5. May participants in clinical trials be compensated?
CTR
The main rule under the CTR is that no undue influence, including that of a financial nature, may be exerted on persons for their participation in the clinical trial. For the categories of incapacitated participants, minors and pregnant women, the CTR prescribes that no incentives or financial inducements be given to the subject or their legally designated representative, except for compensation for expenses and loss of earnings directly related to participation in the clinical trial.
MRA
The MRA follows a similar approach. Participants may be compensated, subject to the following requirements:
- Compensation may not affect the giving of consent to participate in the clinical trial if the subject is under eighteen years of age (Article 3(1)(g) MRA);
- Compensation may not disproportionately affect the giving of consent to participate in the clinical trial if the subject has reached the age of eighteen (Article 3(1)(h) MRA);
- Compensation may not exceed an amount reasonably related to the nature, scope and purpose of the scientific research.
6. How are participants in clinical trials protected and indemnified against any harm that arises as a result of participation in the trial?
The CTR prescribes that Member States must implement a scheme providing for compensation for damage caused by participation in clinical trials within their territory (Article 76 CTR). The Netherlands has opted for compulsory insurance coverage for the conduct of clinical trials to ensure compensation for damage caused by participation in clinical trials. It is compulsory for the person/entity conducting clinical trials in the Netherlands to take out insurance prior to conducting the research (Articles 7 and 8 MRA). Specific requirements for the mandatory taking out of insurance for the conduct of clinical trials in the territory of the Netherlands are set out in the Medical Research (Human Subjects) Compulsory Insurance Decree 2015 (Besluit verplichte verzekering bij medisch-wetenschappelijk onderzoek met mensen 2015). The minimum requirements for the taking out, coverage and amount of the compulsory insurance set out in the Decree may not be derogated from to the detriment of the clinical trial participant (Article 8 of the Decree).
Also from this Legal Handbook
3. Marketing, Manufacturing, Packaging & Labeling, Advertising: The Netherlands
1. What is the authorization process for the marketing of new drugs, biologics, medical devices, non-prescription medications, and other medicinal products?
Please refer to Chapter 1, Question 3.
2. What is the authorization process for the marketing of generic versions of these products?
Please refer to Chapter 1, Question 6.
3. What are the typical fees for marketing approval?
Please refer to Chapter 1, Question 4.
4. What is the period of authorization and the renewal process?
Please refer to Chapter 1, Question 5.
5. What are the requirements, if any, for post-approval pharmacovigilance?
Please refer to Chapter 1, Question 8.
6. Are foreign marketing authorizations recognized?
Medicinal products
MAs granted by the MEB or by the EMA are recognized in the Netherlands. MAs granted by the regulatory authorities in other EU Member States can be recognized through the procedure of mutual recognition (see further Chapter 1, Question 3). MAs from non-EU states are not recognized in the Netherlands.
Medical devices
A manufacturer situated outside the EU has to appoint a sole authorized representative within the EU to be able to put medical devices on the market in the EU. Furthermore, only Notified Bodies within the EU are allowed to perform conformity assessment tasks and grant certificates. A conformity assessment from a Notified Body in one Member State grants the right to trade the medical device throughout the whole EU. An MA from a non-EU authority, such as the FDA, does not release a manufacturer from the obligation to obtain a conformity certificate from an EU Notified Body. However, the evaluating Notified Body can take the findings from the non-EU authority into account in its conformity assessment.
7. Are parallel imports of medicines or devices allowed?
Medicinal products
The parallel import of a medicinal product from another EU Member State or an EEA state into the Netherlands is permitted with a parallel trade authorization from the MEB. The medicinal product should be equivalent to a medicinal product that is already authorized in the Netherlands, including in terms of its pharmaceutical form and preferably also its package size. If the MEB already possesses the required information regarding safety as a result of the first marketing of a medicinal product in the Netherlands on the basis of an MA granted by the MEB, the MEB can grant an MA for the parallel-imported medicinal product according to a relatively simplified procedure (Article 48 Medicines Act).
Medical devices
The parallel import of a medical device from a non-EU Member State into the Netherlands is only permitted if the product is accompanied by a valid EU CE marking and DoC.
8. What are the restrictions on marketing practices such as gifts, sponsorships, consultancy agreements, travel and entertainment, or other incentives for healthcare organizations and individual medical practitioners?
Under Dutch law, inducement (gunstbetoon) is defined as the promise, offering or granting of money or services or goods that can be valued in money with the apparent aim of promoting the prescription, supply or use of a medicinal product or medical device (Article 1(1)(zz) Medicines Act and Article 6(1) Medical Devices Act). The inducement rules apply to all health professions and other persons who or organizations that professionally influence the choice regarding the medicinal product or medical device. This is a wide scope, which not only includes practitioners but also other persons with professional influence on the decision to use a particular medical device, e.g. a nurse or a purchaser at a healthcare institution.
In principle, inducement is prohibited for both medicinal products and medical devices. However, not all forms of inducement are prohibited. For both medicinal products and medical devices, the following exceptions apply (Article 94 Medicines Act and Article 6 Medical Devices Act):
- Providing compensation for the costs of participation in an event or not charging for participation in an event is permitted. However, such compensation must not exceed the amount that is strictly necessary to participate in the event.
- Remuneration for services rendered by healthcare professionals is permitted, provided the amount is reasonable and proportionate. In addition, the services provided must be relevant to the medical device supplier or healthcare professional involved.
- Gifts of a limited value (i.e. less than EUR 50, with a maximum of three gifts per individual healthcare professional per year) are permitted if they are relevant to the healthcare professional’s practice.
- Reduced rates and bonuses relating to the purchase of medical devices are permitted.
The rules on inducement for medicinal products and medicinal devices are explained in more detail in, respectively, the Policy Rules for Inducements under the Medicines Act 2018 and the Policy Rules for Inducements under the Medical Devices Act. These policy rules elaborate on the content and scope of the ‘inducement’, including in particular the exceptions to the prohibition of inducement listed above. The Inspectorate applies these policy rules in the enforcement of, respectively, the provisions on advertising of the Medicines Act and Article 6 of the Medical Devices Act.
Moreover, businesses are obliged to provide data regarding the financial relationship between healthcare professionals and businesses, along with the individual registration number, the BIG number of the healthcare professional, in a public transparency register (Article 13a of the Medicines Act Decree). Whether a relationship between a professional and a business qualifies as such is laid down in the Code to prevent undue influence through conflicts of interest (Code ter voorkoming van oneigenlijke beïnvloeding door belangenverstrengeling).
9. How is the manufacturing of medicines and devices regulated and by which agencies?
Manufacturers must adhere to the GMPs. The Inspectorate is responsible for the monitoring of the GMP standards and conducts inspections of compliance by manufacturers and wholesale distributors.
Medicinal products
The manufacturing of medicinal products and the wholesale distribution of medicinal products in the Netherlands is subject to a manufacturing or wholesale distribution authorization. The application for either type of authorization must be submitted to Farmatec. If the authorization is granted, Farmatec will register the authorization in EudraGMDP, the relevant EMA database.
Medical devices
Manufacturers of medical devices should manufacture the medical device taking into account the requirements for the CE marking.
See further Chapter 1, Question 3.
10. Are local manufacturing requirements compatible with Good Manufacturing Practices (GMPs) as defined by the U.S. Food & Drug Administration and/or the European Medicines Agency?
Yes. The Netherlands has transposed the EU Directives on good manufacturing practices for medicinal products into Dutch law.
11. What is the inspection regime for manufacturing facilities?
The Inspectorate conducts periodic inspections of all manufacturers of medicinal products in the Netherlands, assessing whether the manufacturers comply with GMP and GDP. The Inspectorate usually combines the GMP and GDP inspections.
12. Are manufacturing facilities open for inspection by foreign inspectors or third-party inspectors as authorized by the FDA/EMA?
In principle, the Inspectorate conducts inspections at manufacturing facilities, if necessary in cooperation with the police, customs or the Fiscal Information and Investigation Service (the Fiscale inlichtingen- en opsporingsdienst (FIOD)).
The EU has signed a mutual recognition agreement with the United States that allows the FDA and the EMA to rely on each other’s inspections for products covered, in particular concerning human medicinal products.
13. What are the requirements for storage, packaging, and handling of medicines and devices and their constituent components?
Medicinal products
Storage and handling
The storage of medicinal products requires a wholesale permit (see also Chapter 1, Question 3). The wholesale permit is issued by Farmatec and is also required for the following activities: to have in stock, offer for sale, deliver or export or otherwise bring into or take out of Dutch territory, or operate a wholesale business in medicinal products for which an MA is granted (Article 38 of the Medicines Act).
The rules for storage and distribution are described in the EU Guidelines of 5 November 2013 on Good Distribution Practices (GDP) which give guidance on the obligation for wholesale distributors to appoint a responsible person with responsibility for implementation of and compliance with the GDPs, and on temperature conditions and monitoring, the training of personnel, documentation, the cleaning regime, and the storage of medicinal products or active substances.
To ensure sufficient availability of medicinal products, MA holders and wholesale distributors are obliged to hold sufficient stock of each medicinal product that they hold an authorization for or supply. This requires MA holders to ensure that the medicinal product covered by the MA is sufficiently stocked by wholesalers or pharmacists to meet patients’ needs (Article 49(9) Medicines Act). A wholesale distributor is obliged to hold such an assortment and stock of medicinal products that it can promptly meet the demand for a medicinal product. To further combat medicinal product shortages, as of 1 January 2023, MA holders and wholesale distributors are obliged to keep a ‘base stock’ (‘ijzeren vooraad’) of medicinal products. This base stock is a further interpretation of the obligation to hold sufficient stock and requires the MA holder to hold a stock of at least six weeks. The wholesale distributor should hold at least a two-week stock. This obligation will be extended to a stock of four weeks from 1 July 2023.
Packaging and labeling
The packaging and labeling requirements for medicinal products are set out by the MEB in the policy document MEB 6: Labelling of pharmaceutical products’ (“MEB 6“) and the EU Guideline on the Readability of the Labelling and Package Leaflet for Medicinal Products for Human Use (“Labeling Guideline“).
The information on the packaging must be clearly legible and non-erasable. The information must be presented in (at least) the Dutch language. An exemption from having to use the Dutch language may be granted in exceptional cases laid down in Article 4a.3(2) and (3) Medicines Act Regulation, in which case English is the only permitted alternative language. The product name must also be stated in braille.
The MEB 6 and Labeling Guideline define the items that must be included on the label, including:
- the name of the medicinal product;
- the active ingredient and excipients;
- the dosage strength of the medicinal product;
- pharmaceutical form and contents;
- any special warnings;
- expiration information;
- instructions for use;
- MA holder information; and
- Batch number and Unique Device Identification number (UDI).
For non-prescription medicinal products, the packaging should state the therapeutic indications and the instances in which the medicinal product cannot be used. Based on the rules laid down in the EU Falsified Medicines Directive (2011/62/EU), the packaging of prescription medicinal products should include a unique identifier code and an anti-tampering device, for example a seal on the packaging showing that the package has not been opened.
For more detailed packaging and labeling requirements we refer to policy document MEB 6 and the Labeling Guideline.
Medical devices
The requirements for medical devices are laid down in Annex I of the MDR and IVDR. The information on the packaging must be clearly legible and non-erasable and must include clear instructions for use that are adapted to the level of training and knowledge of the user. Instructions are only not required for class I or IIa medical devices or in some instances for in vitro diagnostic devices, and in the event that the device can be used safely without instructions for use.
The label with the information has to be placed on the device itself. If this is not possible, the information should be placed on the packaging of the device.
The information on the label must be in the Dutch language. However, for medical devices which are exclusively supplied to professional users, the information on the label and instructions for use can be in the English language.
The labeling requirements for medical devices include:
- the name of the medical device;
- necessary details for a user to identify the medical device, the contents of the packaging, and the intended purpose of the medical device;
- the name and address of the manufacturer or authorized representative;
- the lot number or the serial number of the device and Unique Device Identification number (UDI);
- an indication of the date by which the medical device should be used;
- any storage or handling instructions; and
- any warnings or precautions.
For certain medical devices, additional labeling requirements apply. All information and labeling requirements are specified in Annex I of the MDR (Article 23.1 of Chapter III and 23.2) and the IVDR (Article 20.1 and 20.2 of Chapter III). We refer to these articles for more detailed packaging and labeling requirements.
14. What information must be included in medicine and device labeling?
Please refer to Question 13 of this Chapter.
15. What additional information may be included in labeling and packaging?
Medicinal products
The use of signs, images or pictograms, as well as other information which corresponds to the summary of product characteristics (SmPC) by the EMA, which contributes to the health information for a user, is in principle permitted.
The MEB has compiled a list of symbols (listed in Annex 5 of the MEB 6) whose use is permitted. Images may only be used for clarification purposes and may not replace compulsory explanation in text. If signs, pictograms, logos or images are used, they should meet the following requirements:
- the image and text must be coherent;
- the use must correspond with the approved SmPC text and the text cannot be misleading;
- the use must be in line with the Labeling Guideline;
- the visuals must make the text more understandable;
- the use is not contradictory to good taste or decency; and
- the use does not contain any recommendation of the product and cannot be interpreted as any type of promotion of the medicinal product.
The packaging may not include direct or indirect references to (recommendations by) scientists or healthcare professionals.
Medical devices
According to Article 7 MDR, it is prohibited to use text or other signs in the labeling, instructions for use and advertising of devices that may mislead the user with regard to the device’s intended purpose, safety and performance.
16. What items may not be included in labeling and packaging?
Please refer to Question 15 of this Chapter.
17. What are the restrictions and requirements for the marketing and advertising of medicines and devices?
Medicinal products
Direct public advertising is prohibited for prescription-only medicinal products and for products that may be dispensed without a prescription that contain substances as referred to in list I or II of the Opium Act. Advertising for medicinal products without MA or a valid approval number issued by the Inspection Board for the Public Commendation of Medicinal Products (Keuringsraad Openlijke Aanprijzing Geneesmiddelen (KOAG)) is prohibited as well. Direct public advertising of other medicinal products is allowed, provided the relevant requirements are complied with.
The advertising rules for medicinal products are compiled in the Code of Conduct for Pharmaceutical Advertising which elaborates upon the relevant regulations of the Medicines Act and the Dutch Advertising Code (Nederlandse Reclame Code). Of particular relevance is the Code for Advertising Medicinal Products to the General Public 2019, part of the Code of Conduct for Pharmaceutical Advertising (see also Question 1 of this Chapter).
Compliance with the rules for public advertising of medicinal products is monitored by the Inspectorate in cooperation with the Health Advertising Knowledge and Advice Board (Keuringsraad Kennis en Advies Gezondheidsreclame).
Public advertising for any medicinal product must at least contain (Article 8.1 Code for Advertising Medicinal Products to the General Public 2019 (Code Publieksreclame voor Geneesmiddelen (CPG) 2019)):
- the name of the medicinal product;
- the generic name of the active ingredient (if the medicinal product contains only one active ingredient). This requirement does not apply for radio advertisements;
- the indications and contra-indications; and
- an explicit request to read the package leaflet or the text on the outer packaging. In media with space or time constraints, the statement ”please read the information on the package before buying” is considered sufficient, provided the outer packaging of the product contains full instructions for use, or an exhortation to read the package leaflet before use, provided that the statement does not mislead the consumer.
We refer to the Code of Conduct for Pharmaceutical Advertising for more detailed advertising requirements for prescription and non-prescription medicinal products (Articles 5.2 to 5.8).
Medical Devices
Advertisements for medical devices must be conducted by, or on behalf of, the manufacturer or its authorized representative. Advertisements are only allowed for medical devices which are manufactured or distributed in accordance with the Medical Devices Act and the Medical Devices Decree, and for medical devices for the self-care of indications which can be determined by the user without the intervention of a doctor, or which are used for the supplementary self-care for indications determined by a doctor and treated by other means. The rules for advertising of medical devices are laid down in the Code of Conduct Medical Devices and the Code for Advertising for Medical Devices for self-care (Code reclame voor Medische zelfzorg Hulpmiddelen (CMH) 2019).
Advertisements must:
- include the name of the medical device;
- explain the nature of the product;
- contain information that is indispensable/essential for proper use of the medical device;
- be truthful and must not exaggerate the properties of the medical device;
- be in such form and content that they clearly show that they are advertising;
- be in such form and content that the product is clearly recognized as a medical device: the medical device may not equate itself with a medicinal product, health product foodstuff, cosmetic product or other consumer product;
- use language that is understandable to the consumer in terms of medical and scientific terminology; and
- consist of text and presentation that complies with the law and must comply with the prevailing standards of good taste and decency.
We refer to the Code for Advertising for Medical Devices for self-care for more detailed advertising requirements for medical devices for self-care.
18. Where can medicines and devices be sold or delivered? Can medicines and devices be sold or delivered via post?
Medicinal products
The restrictions on sale and/or delivery differ and depend on the type of medicinal product concerned.
The Medicines Act distinguishes between four different types of medicinal products (Article 56 Medicines Act):
- Medicinal products that are prescription-only (‘UR medicinal products’), which may be sold by:
- pharmacists practicing their profession in a pharmacy;
- general practitioners who hold a license to sell;
- persons and bodies designated for that purpose by ministerial regulation.
- Medicinal products that may be dispensed without a prescription, though only in a pharmacy (‘UA medicinal products’), which may be sold by:
- pharmacists practicing their profession in a pharmacy;
- general practitioners who hold a license to sell;
- persons and bodies designated for that purpose by ministerial regulation.
- Medicinal products that may be dispensed without a prescription, though only in a pharmacy or a point of sale supervised by a druggist (‘UAD medicinal products’), which may be sold by:
- pharmacists practicing their profession in a pharmacy;
- general practitioners who hold a license to sell;
- persons and bodies designated for that purpose by ministerial regulation;
- druggists who practice in a drugstore or other point of sale of UAD medicinal products, though under the condition that they offer responsible care. This includes dispensing under the responsibility and supervision of a druggist. A druggist must clearly inform the consumer of what they reasonably need to know about the nature and purpose of the medicinal product and its expected effects and risks for their health, unless they have indicated that they do not require this information. There must be sufficient (assistant) druggists present at the point of sale who can give this information to the customers.
- Medicinal products that may be dispensed without a prescription, which may also be dispensed outside a pharmacy or an outlet supervised by a druggist (‘AV medicinal products’), which may be sold by:
- pharmacists practicing their profession in a pharmacy;
- general practitioners who hold a license to sell;
- persons and bodies designated for that purpose by ministerial regulation;
- druggists who practice in a drugstore or other point of sale of UAD medicinal products, under the condition that they offer responsible care; and
- druggists who conduct sales activities in the course of business and are registered in the trade register for that purpose, under the condition that they offer responsible care.
It is prohibited to remotely offer medicinal products for sale (including online sales) other than in accordance with the Dutch rules implementing Directive 2001/83. For this purpose, there is a national website (only in Dutch) which lists all websites on which medicinal products are legally sold online and delivered by post. These authorized websites must contain the Dutch version of the EU-certification logo for online providers of medicinal products on its website.
Medical devices
The requirements for the sale of medical devices follow from the MDR. Medical devices may be marketed only if they are CE-marked and a DoC for the device has been drawn up. Authorized sales are determined by the specific conditions for market authorization as laid down in the conformity assessment of the specific medical device covered by the CE marking. It follows from the MDR and the IVDR (see e.g. Annex I on the general safety and performance requirements) that in the conformity assessment, a differentiation is made between e.g. medical devices (and in vitro diagnostic medical devices) intended for use by ‘healthcare professionals’ and medical devices intended for use by a ‘lay person’. The conditions for market authorization include conditions based on the intended purpose of the device.
19. What are the restrictions and requirements for electronic marketing and advertising via email, via the Internet, social media, and other channels?
As a starting point, online advertising is subject to the same rules as offline marketing and advertising in the Code of Conduct for Medical Devices and the Code of Conduct for Pharmaceutical Advertising (see further Question 17 above).
Online marketing
For online marketing, the following applies. Dutch providers of medicinal products that (are aiming to) sell medicinal products online through a web shop must register with the CIBG. Following registration, the CIBG will check whether the provider is a legitimate seller that meets the conditions, and include this provider in the overview of web shops offering medicinal products featured on its website. The provider is furthermore required to place the Dutch version of the EU-certification logo for online providers of medicinal products on its website.
Online advertising: social media
Article 5.5.1. of the Code of Conduct for Pharmaceutical Advertising indicates that the following general advertising rules apply to social media advertising:
- advertising shall always be recognizable as such (Article 5.2.1.4);
- the person sending the message or responsible or jointly responsible for its content shall be recognizable (Article 7.1.3);
- addresses must be able to be properly identified and selected. The basic principle is that advertisements are solely addressed to professionals who have an interest in the advertised information (Article 5.8); and
- a responsibility regime applies for the content of own websites and media referred/linked to (see also Article 5.8.12).
Online advertising: direct messaging/email
The legal framework for the advertising of medicinal products consists of the ‘general’ rules for online advertising and the specific rules for the advertising of medicinal products. As regards direct messaging, it is prohibited to send ‘spam’ to consumers. Advertising by direct messaging is only permitted when the following three cumulative requirements are met:
- the recipient has clearly given permission;
- the recipient can confirm that the messages originate from the advertiser’s company;
- the recipient can unsubscribe from the messages easily, online and free of charge.
The ‘Advertising by Email Code’ (Code reclame via e-mail) lists the requirements for advertising by email.
Online advertising: other media
Pursuant to the Media Act (Mediawet 2008), the following restrictions are in place:
- program offerings in public media services may not include advertising and teleshopping messages for medical treatments (Article 2.94 Media Act). Product placements are prohibited (Article 3.88 Media Act).
- program offerings in commercial media services may not include advertising and teleshopping messages for medical treatments (Article 3.7 Media Act).
- product placement for medical treatments is not permitted in commercial media services (Article 3.19 Media Act).
Furthermore, Article 96 Medicines Act prohibits the broadcasting of teleshopping messages for medicinal products in general.
20. May medicines and devices be advertised or sold directly to consumers?
Please refer to Question 17 of this Chapter for information on advertising directly to consumers.
Please refer to Question 18 of this Chapter for information on direct sales to consumers
21. How is compliance monitored?
The Inspectorate monitors websites offering and advertising medicinal products. In particular, the Inspectorate focuses on prescription medicinal products and medicinal products which are not yet registered. The Inspectorate monitors compliance in cooperation with the Health Advertising Knowledge and Advice Board.
The Advertising Code Committee (Reclame Code Commissie) monitors compliance with the different advertising codes.
22. What are the potential penalties for noncompliance?
The Inspectorate may impose sanctions in case of noncompliance and usually imposes a fine, which in principle amounts to EUR 150,000 for large businesses but ultimately depends on the size of the business and the severity of the offense. In some cases, the Inspectorate confines itself to issuing a warning.
The Advertising Code Committee cannot impose any formal measures. However, the committee has a strong informal influence, and its recommendations are usually followed by the party addressed.
Also from this Legal Handbook
4. Traditional Medicines and OTC Products: The Netherlands
1. What are the regulatory requirements for traditional, herbal, complementary, or alternative medicines and devices?
In principle, the regulatory framework set out in the previous Chapters applies. However, differing requirements or procedures apply for specific types of medicinal products. The Medicines Act distinguishes several types of medicinal products, including homeopathic medicinal products and herbal medicinal products. The definitions of these medicinal products correspond to the definitions provided by Directive 2001/83/EC:
- A homeopathic medicinal product is a medicinal product obtained by a homeopathic manufacturing process from raw materials referred to in the homeopathic-pharmaceutical literature as homeopathic raw materials (Article 1(f) Medicines Act).
- A herbal medicinal product is a medicinal product exclusively containing as active ingredients one or more herbal substances or herbal preparations, or a combination thereof (Article 1(i) Medicines Act). Medicinal products that also contain vitamins or minerals complementing the active herbal ingredients mentioned above with respect to the specified indications (Article 1(5) Medicines Act) are also considered herbal medicinal products.
Herbal medicinal products
Like all medicinal products, herbal medicinal products must be authorized by the MEB before they can be marketed in the Netherlands. An abridged procedure applies to traditional herbal medicinal products if the applicant shows that the product fulfills the cumulative conditions of Article 42(8) Medicines Act:
- the product has indications that are exclusively appropriate to a herbal medicinal product that by virtue of its composition and purpose is intended and designed as a medicinal product for use without the supervision of a medical practitioner;
- the product is exclusively intended to be administered in a specified concentration and dosage;
- it is a medicinal product for oral or external use or for inhalation;
- the product or an equivalent or comparable product has been in medicinal use for at least thirty years preceding the date of application for marketing authorization, at least fifteen of which in the European Union;
- sufficient data is available to demonstrate that the product has a traditional use and is harmless; and
- because of its long-standing use and experience, it is plausible that the medicinal product has a pharmacological effect or is otherwise effective.
For traditional herbal medicinal products, the applicant is not obliged to submit clinical and pre-clinical data. Instead, the MEB will assess the efficacy by reference to its long-standing use and experience (Article 42(8)(f) Medicines Act). The MEB will grant authorization for traditional herbal medicinal products if investigation shows the following: the conditions of Article 42(8) Medicines Act are fulfilled, the medicinal product is harmless under normal conditions of use, the medicinal product has pharmacological effects or efficacy, the pharmaceutical quality has been adequately demonstrated, and the product is not a homeopathic medicinal product (Article 45(2) Medicines Act). The MEB also takes into account the Community herbal monographs established by the Committee for Herbal Medicinal Products and, where appropriate, marketing authorizations or similar authorizations granted in another Member State. If the MEB refuses to grant a marketing authorization, it informs the Commission and, upon request, the competent authority of any other Member State.
Non-traditional herbal medicinal products that do not comply with the abovementioned criteria are subject to regular authorization by the MEB.
Homeopathic medicinal products
For the authorization of homeopathic medicinal products a similar abridged procedure as for traditional herbal medicinal products can be followed if the applicant shows that the application concerns a medicinal product that fulfills the following cumulative conditions (Article 42(3) Medicines Act):
- it is a medicinal product for oral or external use;
- no mention of any therapeutic indication is made either in or on its packaging, or in its package insert; and
- there is a sufficient degree of dilution to guarantee the safety of the medicinal product, in any case, the medicinal product may not contain either more than one part per 10,000 of the mother tincture or more than 1/100th of the smallest dose used in allopathy with regard to active substances whose presence in an allopathic medicinal product result in the obligation to submit a doctor’s prescription.
Non-classical homeopathic medicinal products that do not comply with the abovementioned criteria are subject to regular authorization by the MEB.
The Botanicals and Novel Foods (BNV) department of the MEB is responsible for the assessment of an application dossier for a classic and non-classic homeopathic medicinal products and traditional and non-traditional herbal medicinal products. If the medicinal product is positively assessed by the MEB, the manufacturer is granted a marketing authorization. The medicinal product is then entered in the Register of Medicinal Products and a registration number is attributed.
Herbal supplements
Herbal supplements are covered by the Commodities Act (Warenwet). In this regard, the Commodities Act only prescribes that products must be safe for use. The Dutch Food and Consumer Product Safety Authority (Nederlandse Voedsel- en Warenautoriteit (NVWA)) monitors compliance with the Commodities Act, investigates whether food supplements and herbal supplements contain prohibited substances, and checks their production, composition and labeling. Medical claims are prohibited (see further Question 3 of this Chapter).
For (alternative) medical devices, the regulatory framework as set out in previous Chapters applies.
2. Can these traditional, herbal, complementary, or alternative products be advertised directly to the public?
For the general framework applicable to advertising of medicinal products, please refer to Chapter 3, Question 20.
Traditional herbal medicinal products
Article 87 Medicines Act prescribes that advertising must include the following information:
- that a traditional herbal medicinal product is concerned;
- for which indications the product is used; and
- that the indications are based exclusively on a long-standing use.
Homeopathic medicinal products
Article 84(2) Medicines Act with reference to Article 69(1) Directive 2001/83/EC defines a strict scope of information for use in advertising. In addition to the clear mention of the words “homeopathic medicinal product”, the labeling and, where appropriate, the package insert, must bear the following, and no other, information:
- the scientific name of the stock or stocks followed by the degree of dilution, making use of the symbols of the pharmacopoeia,
- name and address of the registration holder and, where appropriate, of the manufacturer,
- method of administration and, if necessary, route,
- expiry date, in clear terms (month, year),
- pharmaceutical form,
- contents of the sales presentation,
- special storage precautions, if any,
- a special warning if necessary for the medicinal product,
- manufacturer’s batch number,
- registration number,
- “homeopathic medicinal product without approved therapeutic indications”,
- a warning advising the user to consult a doctor if the symptoms persist during the use of the medicinal product.
3. What health, advertising, and marketing claims may be made for traditional, herbal, complementary, or alternative products?
The general rule is that medical claims may only be used for medicinal product advertising if they are in line with the registered indication and the information contained in the summary of product characteristics. It is prohibited to make medical claims for products for which the respective party does not hold an MA (Article 84 Medicines Act). For traditional herbal medicinal products and homeopathic medicinal products, the limitations listed above in Question 2 of this Chapter apply to advertising.
The prohibition on medical claims also applies to food products. In this regard, the difference between a medical claim and a health claim can be subtle. A medical claim is regarded as constituting information which attributes to the product the property of preventing, treating or curing a human disease, or the reference to such properties (Article 7(3) of Regulation (EU) 1169/2011).
The presentation of medical claims for products, in particular foodstuffs, may have the consequence that the product in question is regarded as falling under the definition of medicinal product. After all, any substance or combination of substances that is presented as suitable for a medical purpose is regarded as a medicinal product (Article 1(b) Medicines Act). For food stuffs, this prohibition can also be enforced by the Dutch Food Safety Authority. For example, a company which allows for persistent posting of reviews on its website in which medical claims for its product are made, has in the past been considered to have presented the product as suitable for a medical purpose. This was considered to constitute advertising of the product as a medicinal product without possession of the required marketing authorization. There are a limited number of exceptions to the prohibition of medical claims for foodstuffs, such as the exception for food for medical use following from Article 5(2) of Regulation (EU) 2016/128. For these foods, it is mandatory to state the disease, condition or ailment for which the product is intended.
4. What are the regulatory requirements for over-the-counter (non-prescription) medications?
The Medicines Act distinguishes between prescription-only medicinal products (‘UR medicinal products’), and over-the-counter (“OTC“) medicinal products. OTC medicinal products are also referred to as so-called ‘self-care’ medicinal products, since they do not require a prescription. The Medicines Act distinguishes three classes of OTC medicinal products on the basis of the potential risks of the product (Article 56 of the Medicines Act). These three categories are:
- Pharmacy only (UA): medicinal products with a relatively mild potential risk;
- Pharmacy or druggist only (UAD): medicinal products with a relatively low potential risk;
- General Sale (AV): Medicinal products with a relatively very low potential risk.
The MEB determines whether a medicinal product can be sold without prescription and in which class a medicinal product belongs (on the basis of the criteria of Article 57 and 58 Medicines Act). Like all medicinal products, OTC medicinal products are subject to MA. However, depending on the composition of the product concerned, the authorization an abridged procedure may apply (see Question 1 of this Chapter). Compared to prescription-only medications, OTC medicinal products are subject to different regulations with regard to advertising (see further Questions 2 and 6 of this Chapter) and dispensation (see further Question 5 of this Chapter).
5. Are there any limitations on locations or channels through which OTC products may be sold?
The applicable limitations on the locations or channels through which OTC medicinal products may be sold differ according to, and depend on, the type of OTC product.
For the restrictions and requirements on the sale of pharmacy-only (UA) OTC products, pharmacy or druggist-only (UAD) OTC products, and the general sale of (AV) OTC products, please refer to Question 18 of Chapter 3.
6. What health, advertising, and marketing claims may be made for OTC products?
Medical claims for OTC products must be in line with the registered indication and the information contained in the summary of product characteristics. See further Question 3 of this Chapter.
7. Can OTC products be marketed or advertised directly to the public?
Please refer to Chapter 3, Question 17.
8. What is the mechanism by which a prescription-only product can be converted to an OTC product?
The MEB may take a new decision on the classification of a medicinal product if new information is brought to its attention proving that reconsideration is necessary (Article 59 Medicines Act).
9. What are the requirements for the importation of either traditional medicines or OTC products?
The general rule is that MA is required for the import of traditional medicinal products and OTC medicinal products (see further Chapter 1, Question 3).
Also from this Legal Handbook
5. Product Liability: The Netherlands
1. What types of liability are recognized in your jurisdiction?
Dutch liability law generally distinguishes between contractual liability on the one hand and non-contractual liability on the other. Non-contractual liability not only includes fault-based liability (‘onrechtmatige daad‘) but also includes a regime of strict liability for defective products (‘productaansprakelijkheid‘). The regime for strict liability constitutes the Dutch implementation of the EC Product Liability Directive (Council Directive 85/374/EEC, “Directive“).
Contractual liability
A core value of Dutch contract law is the autonomy of contract parties. The starting principle of contract freedom entails the freedom of parties to enter into agreements (or to refrain from doing so) and to determine the content thereof. Dutch contract law is regulated in the Dutch Civil Code (DCC). Contractual liability obviously requires a contractual relationship between the party at fault and the injured party. Article 6:74 DCC provides that a party is liable for damage that the creditor suffers as a result of any shortcoming in the compliance with a contractual obligation, unless the shortcoming is not attributable to the debtor. With regard to purchase agreements, the DCC prescribes that products must possess the qualities that are expected of them, taking into account the nature of the product in question and any statements made by the seller (Article 7:17 DCC). If a product does not possess the qualities agreed, the purchaser can demand certain remedies from the seller, such as delivery of the missing parts of the product, repair or replacement of the product, or a price reduction. A purchaser can also terminate the contract if the seller fails to fulfill the obligation agreed upon by contract (Article 6:265 DCC).
Strict liability for defective products: product liability (Articles 6:185 – Article 6:193 DCC)
Contrary to contractual liability, under the strict liability regime an obligation to compensate damage can also arise without an existing contractual relationship between parties. The Directive imposes strict liability on the manufacturer of a defective product. Product liability relates to damage caused by the product, instead of damage to the product itself. The regime of strict product liability entails that that a seller, distributor or manufacturer of a defective product is liable to a person injured by that product regardless of whether or not that seller, distributor or manufacturer acted intentionally or negligently. A manufacturer of products cannot contractually exclude or limit its strict liability for defective products (Article 6:192 DCC).
See further Question 2 of this Chapter for more information on Dutch product liability law.
Non-contractual: tort-based liability (Article 6:162 DCC)
Whilst a product liability claim can be based on the specific regime of strict liability for defective products, it can also be based on the general tort law regime. A civil wrongdoing between private individuals creates the legal obligation to compensate damage, as prescribed by tort law in Article 6:162 DCC. Tort-based liability requires an unlawful act that is also attributable to a person and that caused certain damage. If attributability cannot be proven by a claimant, a tort-based claim will generally not be awarded. Under Dutch tort law, an unlawful act could be an infringement of rights, an act or omission in breach of a statutory duty, or a breach of what is accepted as customary unwritten law within society. With respect to injuries or property damage, an unlawful act is easily assumed.
- Contractual Liability
Plaintiffs that are party to a contract with a defendant manufacturer may make a claim in contract. In contract law, plaintiffs can enforce express terms or terms implied from legislation, trade practice and custom against defendant manufacturers. Common law provinces have enacted legislation requiring manufacturers to provide warranties of product quality and fitness, and the Civil Code of Québec requires manufacturers to provide a warranty of quality, including a warranty against latent defects. Consumer protection acts across Canada further legislate into consumer contracts implied terms of description, fitness, merchantability, and durability of the product, and provide consumers additional remedies for false, misleading, or deceptive representations.
- Civil Liability
Plaintiffs that are not party to a contract must claim under a civil wrong. Common law plaintiffs can claim under the tort of negligence. Specific claims include negligent manufacture of a product, negligent design of a product, or the failure to warn users of product dangers. Negligence claims require proof that the product was defective, that the defendant owed a duty of care to the plaintiff, that the defendant negligently breached the standard of care with respect to the product, that the plaintiff’s damages were foreseeable, and that the defect caused or contributed to the plaintiff’s damages. Québec plaintiffs can claim under a special liability regime in the Civil Code of Québec that protects against safety defects, such as defects in design, manufacture, or preservation, as well as the lack of sufficient instructions, safety precautions, or warnings. This regime requires no proof of negligence. Plaintiffs need only to prove the existence of the product’s safety defect, the damage suffered and a causal link between the two to hold the manufacturer liable.
2. How do these types of liabilities apply to the manufacturers of medicines and devices?
Medical products (medicinal products as well as medical devices) could harm a patient, in which case the liability consequences for manufacturers follow from the context and relationship between the manufacturer and the patient. In the absence of a contractual relationship between a patient and the manufacturer of medical products, a patient is generally reliant on strict liability for defective products and fault-based tort. As fault-based tort required culpable conduct by the manufacturer, which will be rare and, in any case, very difficult to prove, a patient will generally seek to ground a claim in the strict liability regime.
Manufacturer
A general principle of Dutch product liability law is that a manufacturer is liable for damage caused by a defective product put into circulation (Article 6:185 DCC). Under Dutch law, the term “manufacturer” refers not only to the manufacturer of a finished product but also to the manufacturer of raw materials or of a component part, as well as to any person who presents himself as a manufacturer by affixing his name, trademark or other distinctive sign to the product (Article 6:187(2) DCC).
Any person who imports a product into the EEA to sell, rent, lease or otherwise provide it in the course of his commercial activities is considered to be a manufacturer under Dutch product liability law. In addition Dutch law stipulates that where the manufacturer of a product cannot be identified, each supplier shall be treated as its manufacturer, unless this supplier informs the injured party within a reasonable time of the identity of the manufacturer or of the person who supplied him with the product (Articles 6:187(3) and 6:187(4) DCC).
Defectiveness
A product is considered to be defective if it lacks the level of safety that its users are entitled to expect (Article 6:186 DCC). Whether a product is considered to be defective is determined by all relevant circumstances of the specific situation. In particular, the presentation and the foreseeable use of the specific product are taken into account, as well as the moment the product was marketed. With respect to the latter, legislation effective when a product was put into circulation might provide an indication for defectiveness of the product nowadays. A manufacturer must be aware that his products may not at all times be used as initially intended and that warnings may not be sufficiently taken into account as they should be. To some extent, a manufacturer should therefore anticipate the reasonably foreseeable misuse of his product.
Types of damage
The damage that may be eligible for compensation by the manufacturer of defective products is defined by Article 6:190 DCC. This clause provides an exhaustive list of types of damage for which a manufacturer may be liable, such as certain personal injury or property damage. With regard to personal injury, damage due to death and/or physical injury, the claimant will in principle be compensated by the manufacturer for such damages caused by the injury – assuming the manufacturer is indeed liable. Property damage will generally only be compensated if it was caused to other goods intended for regular use or consumption in the private sphere. Moreover, the defective product must have been used by the claimant primarily in that sphere. Consequential damage can then also qualify for compensation. For property damage, a threshold of EUR 500 applies: only if property damage amounts to more than EUR 500 will a claimant be able to recover his damages from the seller based on the strict liability regime. For property damages below this threshold, a claim based on fault-based tort remains (Article 6:162 DCC).
Medical products
In the Netherlands, a care provider and patient have a contractual relationship based on a medical treatment agreement. Pursuant to Article 6:74 DCC, the care provider would be liable towards the patient if he failed to fulfill his obligations under this medical treatment agreement. In addition, care providers could also be liable for complications caused by any goods used in performing their medical treatment. Damages arising from the use of a medical product are attributable to the care provider if he did not use the medical device in a way that is overall to be expected of a good care provider (Articles 6:75, 6:77 and 7:453 DCC).
If a medical product used in medical treatment is itself defective, the contractual liability of the care provider in his capacity as the product user will shift to the manufacturer (Article 6:173 DCC). The same applies to any contractual liability of the care provider as seller of the medical product (Article 7:24 DCC). Nonetheless, if these medical product are used, chosen or preserved incorrectly by a care provider, the provider himself is liable, as is the healthcare institution (Article 7:463 DCC).
The MDR prescribes that manufacturers of medical devices must have measures in place to provide sufficient financial coverage in respect of their potential liability under the MDR in a manner that is proportionate to the risk class, the type of device and the size of the enterprise (Article 10(16) MDR).
3. Does potential liability extend to the manufacturer only or could claims extend to corporate executives, employees, and representatives?
As a general starting principle, corporate liability is limited to the legal entity that has caused the damage (tort-based liability) or to the legal entity that put the product into circulation (strict liability). Consequently, potential liability claims generally extend to the manufacturer only and not also to corporate executives, employees and/or other company representatives. Only the legal entity of the manufacturer – and in some specific cases also the importer of products into the EEA or the supplier if a manufacturer is not identified (Article 6:187 DCC) – can be liable for defective products.
In theory, corporate executives, employees and representatives could be liable under tort law. This happens rarely and the specific conditions of a matter will be decisive in determining whether there is reason to allow this exceptional ‘piercing of the corporate veil’. Directors or board members could only be personally liable in exceptional cases, for instance when there is a serious reproach, mismanagement of negligence. With regard to defective products, a director might for instance and under specific circumstances be liable for knowingly trading counterfeit products.
Any person who imports a product into the EEA for sale, rental or leasing, or who otherwise provides it in the course of his business, is also qualified as a manufacturer. Such liability is considered to be no different to that of the actual manufacturer of the product. The rationale behind this is to prevent liability being avoided by manufacturers by putting products into circulation anonymously – it is presumed that importers would in fact be able to trace such manufacturers, for instance for recourse claims, in the case of defective products. To this end, for ‘anonymous products’, suppliers can be equated with the manufacturer (Article 6:187 DCC). The liability of a supplier is relevant when the identity of the actual manufacturer cannot be established. In that case, any supplier can be regarded as the manufacturer, unless he informs the injured party of the manufacturer’s or his own supplier’s identity within a reasonable time. Unless the supplier informs the injured party within a reasonable time about the importer or his supplier in the EEA, he is deemed liable for the defectiveness of the product supplied. This highlights the necessity of proper administration by the supplier in order to trace the origin of individual products.
Revision of the Product Liability Directive
On 28 September 2022, the European Commission adopted a proposal to revise the Directive. If all proposed amendments remain, revision of the Directive will lead to a significant expansion of parties that could be held liable for defective products. The proposed Article 7 ensures that there is always an “economic operator” against whom a claim for damages can be brought. For example, where no authorized representative is available, claims can also be made against “fulfillment service providers” providing packaging or similar services. Secondary liability is also expanded to cover not only retailers and distributors, but also – to the extent applicable – online platforms. Article 7 also provides that manufacturers may be held liable for changes they make to products already placed on the market, including software updates or changes caused by machine learning. This includes parties that market refurbished products. It also means, for example, that a manufacturer of a car navigation system can be held liable if an update of the navigation system turns out to be defective and an injured party suffers damage as a result.
4. How can a liability claim be brought?
Dutch law does not provide special procedural rules for bringing product liability claims, other than regular procedural law rules for bringing claims. Claiming damages for defective products is to take place in accordance with the standard procedures set out under the Dutch Code of Civil Procedure.
Claimants can choose several places to bring legal actions, depending on Dutch procedural law as well as private international law. These include the court of the claimant’s own domicile, the court of the place where the manufacturer has its registered office, and the place where the harmful event occurred. Once the claim is brought before the court, the aggrieved party must prove all elements of the claim. This means that, within the Dutch jurisdiction, the injured party will have to prove the damage, the defect in the product and the causal link between the defect and the damage. Under the strict liability regime applicable to manufacturers of defective products, manufacturers of a product put into circulation bear the risk of defectiveness, regardless of their culpability or fault. The injured party, in that case, does not have to prove that the manufacturer would actually be to blame for the defectiveness of the product in question.
The Act on the Settlement of Mass Damages Claims in Collective Actions (‘Wet Afwikkeling Massaschade in Collectieve Actie’) came into force in 2020. This Act allows an exclusive representative to act on behalf of a collective interest group in collective action proceedings. It enables damages to be claimed directly for an entire group under certain conditions, for instance where damage is caused by a defective product.
Revision of the Product Liability Directive
As the Directive is under revision, we consider it relevant to refer in broad terms to upcoming legislative changes too. The currently proposed amendments to the Directive contain significant alleviations of the burden of proof on claimants.
The proposed Article 8 introduces, for example, an obligation for manufacturers to disclose ‘relevant evidence’ in their possession under certain circumstances. The purpose of this provision is to redress any imbalance between the parties’ advantages and disadvantages due to the asymmetric spread of information about a product. A claimant requesting disclosure must have presented sufficient facts and evidence to make the claim for damages ‘plausible’. Paragraph 2 of this article limits the disclosure obligation to evidence that is ‘necessary and proportionate’ to substantiate this claim for damages. The article does not specify when such information is ‘necessary’ or ‘proportionate’, though the explanatory memorandum does offer the rather general comment that, in determining whether disclosure is proportionate, “the legitimate interests of all parties, including those of relevant third parties, should be taken into account.”
In addition to imposing an obligation on manufacturers to disclose information, the revised Directive will introduce several presumptions of evidence – in favor of consumers – that could potentially have far-reaching consequences in individual or class actions. Article 9 of the revised Directive will contain (i) the presumption of defectiveness of a product and (ii) the presumption of a causal link between the defectiveness and alleged damage. A product will be presumed to be defective if, for instance, the manufacturer fails to comply with the obligation to disclose ‘relevant evidence’ pursuant to Article 8. Secondly, defectiveness is presumed if a claimant establishes that the product does not comply with mandatory safety requirements, or that damage was caused by an obvious malfunction of the product under normal use and circumstances. Lastly, a causal link will be presumed if a defective product and the “type of damage caused is generally consistent with the defect in question”.
5. What defenses are available?
A number of defenses can be raised against a product liability claim within the Dutch jurisdiction.
First and foremost, a manufacturer is not liable for damage caused by a defect in his product if:
- the manufacturer did not put the product into circulation;
- it is plausible, given the circumstances, that the defect that caused the damage did not exist when the manufacturer put the product into circulation, or that this defect arose later;
- the product was neither manufactured for sale or for any other form of distribution for an economic purpose of the manufacturer, nor manufactured or distributed in the exercise of his profession or business;
- the defect is a consequence of the fact that the product complies with mandatory government regulations;
- the state of scientific and technical knowledge at the time the manufacturer put the product into circulation made it impossible to detect the existence of the defect;
- for the manufacturer of a raw material or of a component part, the defect is caused by the design of the product of which the raw material or the component part forms a part, or by the instructions given by the manufacturer of the product.
The “development risk” defense, i.e. that the defectiveness of a product was not discoverable according to the state of scientific and technical knowledge at the time the manufacturer put the product into circulation, rarely leads to successful rebuttal of claims invoked in the Netherlands, or in the rest of Europe. The fact that the defense rarely appears in case law does not, however, mean that it has no practical significance. Many cases in which the ‘development risk defense’ may play a part are settled out of court.
Revision of the Directive
The proposed amendment to the Directive adds an additional defense by which product liability can be avoided. It concerns products that were initially placed on the market legally but are subsequently modified – refurbished – by different manufacturers, which entails that such actions take place outside the original manufacturer’s control.
Limitation period
Under Dutch law, the limitation period for a claim based on the strict product liability regime is three years starting from the moment the damage, the defect and the manufacturer are known or could have been known (Article 6:191 DCC). The DCC also provides a longstop period: in any case, the claim for damages expires 10 years after the product has been put into circulation, as does the right of recourse of a third party (who can also be held liable). Furthermore it is emphasized that if a product consists of separate parts that were put into circulation at different times, the limitation period will start when the last part is put into circulation.
In addition to specific product liability limitation periods, the regular limitation period of five years for tort claims (Article 3:310 DCC) is simultaneously applicable. Both periods apply equally. Consequently, after four years, for instance, a claimant could still bring the same claim before the court, albeit on one legal ground, i.e. tort, and no longer on both tort and product liability.
Also from this Legal Handbook
6. Patents & Trademarks: The Netherlands
1. What are the basic requirements to obtain patent and trademark protection?
Patents
The three main criteria for obtaining patent protection are novelty, inventive step and industrial application (Article 2 Dutch Patent Act (Rijksoctrooiwet 1995), “DPA“).
Trademarks
The basic requirements for obtaining trademark protection are that of a distinctive character of the sign, a clear and accurate representation of the sign in the register, availability in the register (the sign cannot have been registered before by a third party) and the absence of absolute and relative grounds of refusal (see Question 3 of this Chapter).
2. What agencies or bodies regulate patents and trademarks?
Patents
There are several routes to obtaining patent protection for the Dutch territory:
- Filing a Dutch national application with the Netherlands Patents Office (Octrooicentrum Nederland, “OCNL“).
- Filing a European patent application with the European Patent Office (“EPO“).
- Filing an international patent application in accordance with the Patent Cooperation Treaty (“PCT“). The OCNL, the EPO and the World Intellectual Property Organization (“WIPO“) are receiving offices for the PCT application.
On 1 June 2023, the Agreement on a Unitary Patent Court (“UPCA“) has entered into force, and as of that date it is possible to obtain a European Patent with ‘unitary effect’. The Unitary Patent has effect in all EU Member States that participate in the unitary system, (currently 17 countries, including the Netherlands). The Unified Patent Court (“UPC“) will have exclusive jurisdiction over all Unitary Patents. It will also have jurisdiction over European Patents that are validated in participating Member States, unless the patent holder has decided to opt out of the unitary system. It is possible to opt out during the transitional period of 7 years starting 1 June 2023, which period can be extended once with 7 years. The UPC will have a central division, with seats in Paris, Munich and Milan, and various local and regional divisions. The Netherlands will have a local division that will be based in The Hague.
Trademarks
There are several routes to obtaining a trademark for the Dutch territory:
- Filing a Benelux trademark application with the Benelux Office for Intellectual Property (“BOIP“). This is the equivalent of a national trademark for the territory of Belgium, the Netherlands and Luxembourg;
- Filing an EU trademark application with the European Union Intellectual Property Office “EUIPO“);
- Filing an international trademark application with the BOIP, in accordance with the Madrid Agreement and Protocol.
3. What products, substances, and processes can be protected by patents or trademarks and what types cannot be protected?
As a starting point, inventions may be patented within all areas of technology. These can also include inventions relating to a product consisting of or containing biological material, or relating to a process by which biological material is obtained, processed or used, for instance (Article 2a DPA):
- biological material isolated or obtained from its natural environment by means of a technical process, even if such material is available in nature;
- a part of the human body isolated or otherwise obtained by means of a technical process, including a sequence or partial sequence of a gene, even if the structure of that part is identical to that of a natural part;
- plants or animals, provided that the practicability of that invention is not technically limited to a particular plant or animal variety;
- a microbiological or other technical process by which biological material is obtained, processed or used, or a product obtained thereby.
Certain subject matters are inherently not patentable. The DPA provides the following examples of subject matter that will not be regarded as patentable inventions (Article 2 DPA):
- discoveries, scientific theories and mathematical methods;
- aesthetic creations;
- schemes, rules or methods for performing mental acts, playing games or business methods or computer programs;
- presentations of information.
Other exclusions of patentability include (Article 3 DPA):
- inventions for which the commercial exploitation would be contrary to the public order or morality;
- the human body at the various stages of its formation and development, as well as the mere discovery of any of its parts, including a sequence or partial sequence of a gene;
- plant or animal varieties;
- processes of an essentially biological nature, consisting wholly of natural phenomena such as crossing or selection, for the production of plants or animals as well as products obtained thereby;
- inventions in violation of Articles 3, 8(j), 15(5) and 16(5) of the Convention on Biological Diversity; and
- methods of treatment of the human or animal body by surgical intervention or medical treatment and methods of diagnosis applied to the human or animal body, with the exception of products, in particular substances or compositions, for the application of any of these methods.
Articles 4(5) and 4(6) DPA provide for the patentability of substances and compositions that belong to the state of the art that are intended for a new medical use.
Trademarks
The basic requirements are the following (Article 2.1 of the Benelux Convention on Intellectual Property):
- the sign must be a sign, which can include words, personal names, designs, letters, numerals, colors, shapes of goods or packaging of goods, or sounds;
- the sign must serve to distinguish the goods or services of one undertaking from those of other undertakings (either inherently or through use); and
- the sign must be capable of being represented in the register in a manner which enables the competent authorities and the public to determine the clear and precise subject matter of the protection afforded to its proprietor.
Marks can be refused protection on absolute and on relative grounds.
The absolute grounds of refusal are (Article 2.2bis of the Benelux Convention on Intellectual Property):
- signs which cannot constitute a trademark;
- trademarks which are devoid of any distinctive character;
- trademarks which consist exclusively of signs or indications which may serve, in trade, to designate the kind, quality, quantity, intended purpose, value, geographical origin, or the time of production of the goods or of rendering of the service, or other characteristics of the goods or services;
- trademarks which consist exclusively of signs or indications which have become customary in the current language or in the bona fide and established practices of the trade;
- signs which consist exclusively of the shape, or another characteristic, (i) which results from the nature of the goods themselves, (ii) which is necessary to obtain a technical result, or (iii) which gives substantial value to the goods;
- trademarks which are contrary to public policy or to accepted principles of morality in one of the Benelux countries;
- trademarks which are of such a nature as to deceive the public, for instance, as to the nature, quality or geographical origin of the goods or services;
- trademarks which have not been authorized by the competent authorities and are to be refused or invalidated pursuant to Article 6ter of the Paris Convention;
- trademarks which are excluded from registration pursuant to Union legislation or the internal law of one of the Benelux countries, or to international agreements to which the European Union is party or which have effect in a Benelux country, providing for protection of designations of origin and geographical indications;
- trademarks which are excluded from registration pursuant to Union legislation or international agreements to which the European Union is party, providing for protection of traditional terms for wine;
- trademarks which are excluded from registration pursuant to Union legislation or international agreements to which the European Union is a party, providing for protection of traditional specialties guaranteed; or
- trademarks which consist of, or reproduce in their essential elements, an earlier plant variety denomination registered in accordance with Union legislation or the internal law of one of the Benelux countries, or international agreements to which the European Union is party or which have effect in a Benelux country, providing protection for plant variety rights, and which are in respect of plant varieties of the same or closely related species.
The relative grounds of refusal are (Article 2.2ter of the Benelux Convention on Intellectual Property):
- the trademark is identical with an earlier trademark, and the goods or services for which the trademark is applied for or is registered are identical with the goods or services for which the earlier trademark is protected;
- because of its identity with, or similarity to, the earlier trademark and the identity or similarity of the goods or services covered by the trademarks, there exists a likelihood of confusion on the part of the public; the likelihood of confusion includes the likelihood of association with the earlier trademark.
- If it is identical with, or similar to, an earlier trademark irrespective of whether the goods or services for which it is applied or registered are identical with, similar to or not similar to those for which the earlier trademark is registered, where the earlier trademark has a reputation in the Benelux territory (for Benelux trademarks), or in the European Union (for EU trademarks), and the use of the later trademark without due cause would take unfair advantage of, or be detrimental to, the distinctive character or the repute of the earlier trademark;
- an agent or representative of the proprietor of the trademark applies for registration thereof in his own name without the proprietor’s authorization, unless the agent or representative justifies his action;
- in the case of protection of designations of origin and geographical indications:
- an application for a designation of origin or a geographical indication had already been submitted in accordance with Union legislation or the internal law of one of the Benelux countries prior to the date of application for registration of the trademark or the date of the priority claimed for the application, subject to its subsequent registration;
- that designation of origin or geographical indication confers on the person authorized under the relevant law to exercise the rights arising therefrom the right to prohibit the use of a subsequent trademark.
4. How can patents and trademarks be revoked?
Patents
Pursuant Article 75 DPA, a patent will be invalidated by the competent court, if:
- the subject-matter was not patentable (see further Questions 1 and 3 of this Chapter);
- the patent specification does not contain a description of the invention that is sufficiently clear and complete to allow a person skilled in the art to use the invention;
- the subject-matter of the patent extends beyond the content of the (original) patent application;
- the scope of protection has been extended after the patent is granted; or
- the patent holder was not entitled to the patent.
Trademarks
A trademark can be revoked by the competent court, if:
- the proprietor surrenders its registration (Articles 2.25 and 2.26 of the Benelux Convention on Intellectual Property);
- the validity period of ten years expires, if the proprietor does not pay the renewal fee (Article 2.26 of the Benelux Convention on Intellectual Property);
- the trademark has become the common name in the trade for a product or service in respect of which it is registered, though only if that situation occurred as a result of acts or inactivity of the proprietor (Article 2.27 of the Benelux Convention on Intellectual Property);
- if the trademark is liable to mislead the public as a result of its use by the proprietor or with the consent of the proprietor, particularly as to the nature, quality or geographical origin of the goods or services for which it is registered (Article 2.27 of the Benelux Convention on Intellectual Property); or
- if the trademark has not been put to genuine use for five consecutive years for the goods or services for which the trademark has been registered (Articles 2.27 and 2.23bis of the Benelux Convention on Intellectual Property);
- if the trademark is declared invalid on the basis of the absolute and/or relative grounds of refusal in Articles 2.2bis and 2.2ter of the Benelux Convention on Intellectual Property.
5. Are foreign patents and trademarks recognized and under what circumstances?
Foreign patents and trademarks (other than Benelux and EU trademarks and Unitary Patents) are not recognized in the Netherlands. Pursuant to the principle of territoriality, they solely have legal effect in the jurisdictions where they are granted and in force.
It is of note that foreign trademarks can have legal effect in the Benelux if they can be considered ‘well-known’ trademarks in accordance with Article 6bis of the Paris Convention (Article 2.2ter under 2d and 2.19 of the Benelux Convention on Intellectual Property).
6. Are there any non-patent/trademark barriers to competition to protect medicines or devices?
Yes, for example the use of supplementary protection certificates, the tort law doctrine of ‘slavish imitation’, and the Dutch legal framework for protection of trade secrets.
- Supplementary protection certificates
Supplementary protection certificates (“SPC“) provide an additional period of protection after the expiry date of a patent relating to a medicine or active ingredient. This arrangement is put in place to compensate for the period of time in which patent protection was already applied for, but MA had not yet been obtained. An SPC grants the same rights as a national patent and is subject to the same limitations and obligations (article 5 EU regulation 1768/92 concerning the creation of an SPC for medicinal products (“SPC Regulation“)). SPCs are valid for a maximum of five years, depending on the duration of the period between the patent application and MA (Article 13 SPC Regulation). Where it concerns medicines for children for which a pediatric investigation plan has been prepared, the SPC may once be extended by six months. An SPC is granted to the patent holder or his successor in title (Article 6 SPC Regulation) and takes effect at the expiry date of the patent. SPCs are issued by the OCNL (Article 91 DPA) and are subject to annual fees (Article 92 and 95 DPA).
In order to obtain an SPC, the following conditions must be satisfied (article 3 SPC Regulation):
- the product is protected by a basic patent in force;
- a valid MA has been granted;
- the product has not already been the subject of an SPC; and
- the MA is the first is the first authorisation to place the product on the market as a medicinal product.
Any person may bring an action for a declaration of invalidity of the SPC, in case the requirements of article 3 SPC Regulation are not fulfilled, in case the patent has lapsed before its lawful term expires, or in case the patent is revoked or limited to the extent that the product for which the SPC was granted would no longer be protected (article 15 SPC Regulation).
Production exception
An exception to the SPC applies for companies other than the SPC holder, which manufacture medicines destined for a country outside the EU, in which country patent protection does not apply. These manufacturers may manufacture the medicine during the SPC period. Furthermore, during the last six months of an SPC, other manufacturers may manufacture and store such medicine, which facilitates the introduction of the medicine by this company on the EU market immediately after the SPC has expired.
- Slavish Imitation (tort law)
Slavish imitation protects against direct imitation of the appearance of a product. Even though it is not prohibited to use another’s efforts, insight or knowledge disclosed in a product for one’s own benefit and possibility to the detriment of a competitor, such imitations are not allowed if another course of action could have been taken in certain respects – without impairing the soundness and usefulness of the product – and the imitation needlessly causes confusion.
- Trade secrets
The Trade Secrets Protection Act (Wet Bescherming Bedrijfsgeheimen (“Trade Secrets Act“)) provides a legal framework for the protection of trade secrets. Trade secret protection can be granted for any information meeting the following requirements (Article 1 Trade Secrets Act).
The information:
- is secret in the sense that it is not, either in its entirety or in the proper composition and arrangement of its components, generally known to or readily accessible to those within the circles ordinarily concerned with such information;
- has commercial value because it is secret; and
- is subject to reasonable measures, given the circumstances, by the person lawfully in possession thereof to keep it secret.
The Trade Secrets Act offers protection against unlawful acquirement. A trade secret is acquired unlawfully (without consent of the trade secret holder) if it is obtained by means of unauthorized access to or unauthorized appropriation or copying of documents, objects, substances, materials or electronic files lawfully in the possession of the trade secret holder that contain the trade secret or from which the trade secret may be derived, and/or by means of conduct which, given the circumstances, is considered contrary to fair trade practices (Article 2(1) Trade Secrets Act).
The Trade Secrets Act furthermore offers protection against unlawful use or disclosure (without consent of the trade secret holder) by a natural or legal person, unless the use or disclosure is permitted or required by law (Article 3 Trade Secrets Act), if that person (Article 2(2) Trade Secrets Act):
- unlawfully obtained the trade secret;
- breaches a confidentiality agreement or other obligation not to disclose the trade secret; or
- breaches a contractual or other obligation to restrict the use of the trade secret.
The acquirement, use or disclosure is also unlawful if a natural or legal person knew, or, given the circumstances, should have known, that the trade secret was obtained, directly or indirectly, from another natural or legal person who used or disclosed the trade secret in an unlawful manner (Article 2(3) Trade Secrets Act). Similarly, the production, offering or marketing of infringing goods, or the import, export or storage of infringing goods for such purposes, is also considered an unlawful use of a trade secret if the person performing those activities knew or should have known that the trade secret was being used unlawfully (Article 2(4) Trade Secrets Act).
Unlawful acquirement does not occur in the following situations (Article 3 Trade Secrets Act):
- Independent discovery or independent design;
- Observation, examination, disassembly or testing of a product or object which has been made available to the public or which is lawfully in the possession of the person acquiring the information and who is not bound by a legal obligation to restrict the obtaining of the trade secret;
- Exercise of the rights of employees or their representatives to information and consultation in accordance with the law;
- Any other practice which, in the circumstances, is consistent with fair trade practices.
- If the acquirement is permitted or required by the law.
Exceptions to protection are listed in Article 4 of the Trade Secrets Act, and include the following situations:
- exercise of the right to freedom of expression and information pursuant to Article 11 of the Charter of Fundamental Rights of the EU, including respect for media freedom and pluralism;
- disclosure of misconduct, wrongdoing or illegal activities, provided that the infringer was acting for the protection of the public interest;
- disclosure of the trade secret by employees to their representatives in the legitimate exercise of their representative functions in accordance with the law, provided that such disclosure was necessary for such exercise;
- acquirement, use or disclosure for the purpose of a legitimate interest protected under the law.
7. Are there restrictions on the types of medicines or devices that can be granted patent and trademark protection?
Please refer to Question 3 of this Chapter.
8. Must a patent or trademark license agreement with a foreign licensor be approved or accepted by any government or regulatory body?
There is no supervision of license agreements by any government or regulatory body. However, it should be borne in mind that a license only has effect vis-à-vis third parties when the license is registered in the public register held by the relevant IP Office (Article 56(2) of the DPA, Article 2.33 of the Benelux Convention on Intellectual Property).
Also from this Legal Handbook
7. Regulatory Reform: The Netherlands
1. Are there proposals for reform or significant change to the healthcare system?
Reform of the Drug Reimbursement System
The policy on medicinal products of the Ministry of Health Welfare and Sport is based on three core values: quality, accessibility and affordability. In order to improve the affordability of healthcare, the Minister proposed a reform of the Drug Reimbursement System resulting in saving € 140 million per year (see further Chapter 1, Question 13 on its general functioning). However, based on the reactions to the proposal, the Minister informed the House of Representatives on 17 May 2023 of its withdrawal of the proposal because of the potential negative impact thereof on the accessibility of medicinal products. The main criticism to the proposal is that, if manufacturers would not reduce their prices in line with the proposed lowered reimbursement limit, this would result in an additional payment for the patient or a forced switch to a different medicinal product that does fall below the reimbursement limit. Moreover, due to the interaction of the Drug Reimbursement System, the Medicine Prices Act and the preference and purchasing policies of health insurers, the proposal could have a negative impact on the security of supply of medicinal products if manufacturers would only keep the fully reimbursed products on the Dutch market.
Nevertheless, the Minister indicated to continue working on a reform of the Drug Reimbursement System other than via a reduction of the reimbursement limits and will inform the House of Representatives early 2024. Later this year, the Minister will follow up on the interim changes to ensure the functioning of the Drug Reimbursement System.
Transparency Register for Healthcare
As mentioned in Chapter 1 (see Question 1), a Healthcare Transparency Register exists and is used healthcare providers and industry to disclose their financial relationships. Although this central register is not strictly based on obligations of binding law, it gives effect to codes of conduct to which the industry generally is part of.
In March 2020, a legislative proposal to lay down in law a Transparency Register for Healthcare was submitted to the House of Representatives (Tweede Kamer). This register would be introduced by amending the Medicines Act, the Medical Devices Act and the Individual Healthcare Professions Act to include the existence of a mandatory public register. Every transaction between manufacturers and doctors of EUR 50 or more would have to be registered. The aim is to ensure that, in the future, patients can be assured that medicinal products or devices are prescribed because they are the best medical/treatment option for them at the time.
The Council of State (Raad van State) issued advice on the legislative proposal in February 2021, criticizing the proposal for not making sufficiently clear why it is necessary to make the existing Transparency Register mandatory to improve its functioning. In June 2021, the Dutch Data Protection Authority (Autoriteit Persoonsgegevens) issued advice to the House of Representatives, also criticizing the proposal. It noted that a registration in the Transparency Register is a considerable intrusion on the privacy of the individual practitioner. The Authority doubts whether the transparency of the register is necessary and whether access to the register could not be limited to the Inspectorate. The current status is that the proposal is being amended by the petitioner, following these criticisms. It is unclear when the amended proposal will be submitted to the House of Representatives for consideration.
2. When are they likely to come into force?
Please refer to Question 1 of this Chapter above.
Also from this Legal Handbook
8. Cannabinoid Drugs, Medicinal Cannabis and Opioid Drugs: The Netherlands
Cannabinoid Drugs
1. Are Cannabinoid Drugs authorized in your country?
In principle, the Dutch Opium Act (Opiumwet) (“DOA“) prohibits selling or warehousing, as well as manufacturing, preparing, processing, delivering, dispensing and transporting of products included in ‘List I‘ and ‘List II‘ of the DOA. This includes cannabis: the product ‘hemp’ is included in list II and is defined as “any part of the plant of the genus Cannabis (hemp), from which the resin has not been removed, with the exception of seeds“. Hemp oil is included in list I. The DOA defines ‘hemp oil’ as “a concentrate of plants of the genu Cannabis (hemp) obtained by extraction of hemp or hashish, whether or not mixed with oil“.
The above does not apply to medicinal products with cannabinoid drugs, i.e. products containing cannabinoids as active substance. Since such products qualify as medicinal products, they may be authorized in the Netherlands in accordance with the general framework for medicinal products. Please refer to Cannabinoid Drugs, Question 3 for the authorization procedure.
2. What are the regulatory authorities with jurisdiction over Cannabinoid Drugs?
Since the regulatory framework applicable to medicinal products similarly applies to cannabinoid medicinal products, please refer to Chapter 1, question 1 of Exhibit A for the relevant regulatory authorities. Please note that the regulatory authorities in relation to medicinal cannabis do differ in certain respects. In that regard, please refer to Medicinal Cannabis, Question 2.
3. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Cannabinoid Drugs?
Please refer to Cannabinoid Drugs, Questions 12 and 13 on, respectively, the pricing and the reimbursement of medicinal products in general.
Medicinal products with cannabinoids as active substances can only be reimbursed under the basic healthcare insurance coverage (see further Cannabinoid Drugs, Question 10) in the specific instance that they are prescribed for an insured person using cannabidiol as adjuvant therapy for seizures associated with Lennox-Gastaut syndrome (LGS) or Dravet syndrome (DS) in combination with clobazam in patients aged two years and older.
Regarding the pricing and reimbursement of medicinal cannabis, please refer to Medicinal Cannabis, Question 3.
4. Which are the cannabinoid drugs that have received market approval to date?
Medicinal products with cannabinoids as active substances are included in the ‘pharmacotherapeutic compass’ (Farmacotherapeutisch Kompas) of the National Health Care Institute. This register is available online and provides information on the medicinal products for human use which are available in the Netherlands. It provides an overview of medicinal products and medicinal product groups categorized by active substance. In this regard, the pharmacotherapeutic compass includes the medicinal product Epudyolex, an antiepileptic containing cannabidiol as an active substance.
5. Who can prescribe Cannabinoid Drugs?
Which persons may prescribe medicinal products, including those containing cannabinoids as active substances, is regulated by the Individual Healthcare Professions Act (BIG Act); please refer to Cannabinoid Drugs, question 14.
Please see Medicinal Cannabis, Question 8 regarding who may prescribe Medicinal Cannabis in the Netherlands.
6. Is there a list of doctors authorized to prescribe Cannabinoid Drugs?
No; please refer to Cannabinoid Drugs, Question 14.
7. What approvals or notifications are required to prescribe Cannabinoid Drugs?
No; please refer to Cannabinoid Drugs, Question 14.
Regarding Medicinal Cannabis, please see Cannabinoid Drugs, Question 21.
8. Which organizations are authorized to sell/distribute Cannabinoid Drugs available?
Please refer to Opioid Drugs, Question 18.
9. Is there a list of retailers/distributors authorized to sell Cannabinoid Drugs?
No.
10. Are there proposals for reform or significant change to the regulation of Cannabinoid Drugs?
No.
11. When are they likely to come into force?
N/A.
Medicinal Cannabis
1. Is Medicinal Cannabis authorized in the country?
Medicinal Cannabis is not registered as a medicinal product and therefore falls under the scope of the DOA. The DOA provides for the possibility to apply for a DOA exemption for activities involving substances referred to in List I and List II of the DOA (“Opium Exemption“) (Articles 6 and 8 DOA). An Opium Exemption is granted for a fixed period of up to five years and for specific activities. It may be granted for any of the following purposes: public health, animal health, scientific or analytical-chemical research, instructional purposes and trade-related purposes (Article 8 DOA). Renewal is possible but requires a new assessment.
For medicinal cannabis, a specific application process for an Opium Exemption applies. See further Question 3 of this Chapter.
2. What are the regulatory authorities with jurisdiction over Medicinal Cannabis?
Ministry of Health, Welfare and Sport
The Ministry of Health, Welfare and Sport, and specifically the Minister thereof (the “Minister“), is responsible for granting Opium Exemptions under the DOA. An Opium Exemption is required for companies or bodies wishing to carry out activities involving substances referred to in List I and List II of the DOA.
Office of Medicinal Cannabis (Bureau Medicinale Cannabis) (“BMC“)
The Minister has mandated the granting of Opium Exemptions to the BMC. On behalf of the Ministry, the BMC processes and assesses applications for an Opium Exemption relating to cannabis, cannabis resin or its preparations.
The BMC is established in accordance with the requirements of the 1971 Single Convention on Psychotropic Substances. This Treaty provides that a party (in this case the Netherlands) that allows for the cultivation of cannabis or cannabis resin must establish a government office for that purpose. In addition to its monitoring role (granting Opium Exemptions), the BMC is responsible for the cultivation of sufficient medicinal cannabis for scientific research and for the production of medicinal products.
The Inspectorate
The Inspectorate supervises all holders of an Opium Exemption. The BMC submits an application for an Opium Exemption to the Inspectorate for advice. Based on this advice, the BMC decides on the application on behalf of the Minister.
MEB
The MEB provides scientific advice to research parties and is involved in the European discussion on a monograph for Medicinal Cannabis.
Farmatec
Pharmaceutical companies that import and/or export Opium Act substances must apply to Farmatec for an exemption.
3. What is the regulatory framework for the authorization, pricing, and reimbursement of Medicinal Cannabis?
Authorization
Specifically for Medicinal Cannabis, an Opium Exemption (see further Question 2 of this Chapter) may be granted or renewed if the applicant requires such exemption for the cultivation of cannabis, pursuant to an agreement with the Minister (Article 8(2) DOA). Such an exemption for the cultivation of Medicinal Cannabis will only be granted as long as the manufacturer is contracted by the BMC.
Pricing
The prices for Medicinal Cannabis are publicly available on the website of the BMC (only in Dutch). The cost is EUR 27.50 for 5 grams as of 1 September 2021.
Reimbursement
In principle, medicinal cannabis is not reimbursed under the basic healthcare insurance coverage, although there are some health insurers which consider reimbursement as a goodwill gesture in individual cases. Patients therefore need to inquire with their health insurer about medicinal cannabis reimbursement. However, a letter dated 30 May 2022 from the Minister to Parliament indicated that the ‘ex gratia reimbursement’ by health insurers is almost nil. In addition, after advice issued by the National Health Care Institute in 2017 stating that there was insufficient scientific evidence for pain relief or improvement in quality of life, the Minister did not see any reason to change the reimbursement status of Medicinal Cannabis.
The cost of Medicinal Cannabis available at pharmacies can be deducted from patients’ tax returns as a special healthcare expense.
4. How is the production and import of Medicinal Cannabis regulated and by which agencies/authorities?
Production and supply
The production and supply of cannabis for medicinal and scientific purposes is the sole responsibility of the BMC as mandated agency by the Ministry of Health, Welfare and Sport. The BMC supplies pharmacies in the Netherlands but can also supply institutions and agencies abroad if authorized to do so by the Dutch government and the authorities of the countries in question.
The production and supply take place through a closed production chain (under the responsibility of BMC). The system is as follows:
Grower
The BMC contracts the grower through a European tender procedure in which anyone can participate. Strict requirements are imposed on the grower, such as standardized cultivation according to Good Agricultural Practices (GAP) and Good Manufacturing Practices (GMP) guidelines, financial stability and no criminal history. The grower must hold an Opium Exemption and sign an agreement with the BMC (Article 6 and 8 DOA). The grower may grow only what is ordered by the BMC. The entire harvest is bought and physically taken away from the grower. The grower is monitored by the BMC and the IGJ. This procedure is laid down in detail in the Policy rules on opium law exemptions (Beleidsregels opiumwetontheffingen).
The BMC currently has contracted only one producer of medicinal cannabis (‘Bedrocan’), on the basis of a European tender procedure. To ensure the availability of medicinal cannabis, and due to the vulnerability involved in relying on only one producer, the BMC launched a European tender in 2019 to contract two producers. On 6 March 2023, the Minister gave an update on this tender in a letter to Parliament, stating that BMC anticipates that the tender procedure will be fully completed by the end of the second quarter of 2023. The current contractor, Bedrocan, will remain BMC’s grower at least until the new framework agreement enters into effect.
Laboratory
The BMC has contracted an external laboratory through a European tender procedure. A sample of each cannabis harvest (taken by a BMC employee) is sent to the lab. This sample must meet the requirements as laid down in the BMC’s ‘Monograph Cannabis Flower’. The lab is inspected by the Inspectorate.
Packer
The packer is also contracted through a European tender procedure. The contract includes both the task of packaging and logistics services. Each harvest is taken away from the grower in bulk bags after a BMC employee has checked the product and weighed all bags. These bulk bags are repacked at the packer into 5-gram jars. A BMC employee performs checks on this to ensure that the jars are properly weighed and to determine the total packed weight of medicinal cannabis. The packer is inspected by the BMC and the Inspectorate.
Logistics service provider
The logistics service provider is also contracted through a European tender procedure. It takes orders from pharmacists and ensures delivery of the product within 24 hours. The logistics service provider also takes care of invoicing the pharmacists. It is audited by CIBG, BMC and Inspectorate.
Pharmacies
The pharmacy takes a patient’s prescription and ensures delivery to the patient. Pharmacies are monitored by Inspectorate.
Import
The BMC also has a legal monopoly (i.e. the exclusive right) regarding the import of cannabis, cannabis extracts extracts and cannabis resin, so as to prevent cannabis from entering illegality.
5. What approval or notifications are necessary to produce or import Medicinal Cannabis?
To be able to produce Medicinal Cannabis, a contracted producer must have an Opium Exemption (see further Chapter 8, Question 2 and Chapter 9, Question 13).
The exemption application is submitted to the Inspectorate for advice. Partly based on this advice, the BMC makes a decision on the application on behalf of the Minister. The application fee for an Opium Exemption is EUR 1,000. In addition, an annual fee of EUR 700 applies.
Pharmacy exempt from DOA for Medicinal Cannabis
The conditions for the exemptions are laid down in the Dutch Opium Act Decree (Besluit Opiumwet) (“DOA Decree“). The exemption for pharmacists and dispensing physicians applies to the designated substances included in the annexes to this Decree. These designated substances include all List II substances excluding hashish.
6. What is the regulatory framework for the marketing and distribution of Medicinal Cannabis?
Article 85 Medicines Act prohibits any public advertising for medicinal products that contain substances as referred to in List I or List II of the DOA (see further Opiod Drugs, Question 17).
The distribution of Medicinal Cannabis for medicinal purposes by pharmacies is allowed on the basis of Article 5(1)(a) DOA.
7. How can patients obtain Medicinal Cannabis?
Medicinal Cannabis can only be obtained on prescription from a general practitioner. It is mainly prescribed when common treatments and registered medicinal products do not have the desired effect or have too many side effects. The BMC has published leaflets on its website containing a list of conditions for which data has shown that Medicinal Cannabis can be effective, such as epilepsy and chronic pain.
Medicinal Cannabis
Currently, five medicinal cannabis products are available in pharmacies. These are Bedrobinol, Bedrocan, Bediol, Bedica and Bedrolite.
8. Who can prescribe Medicinal Cannabis?
A general practitioner may prescribe Medicinal Cannabis if common treatments and registered drugs do not have the desired effect or cause too many side effects. It is up to the general practitioner to determine in which situation and for which condition Medicinal Cannabis is an appropriate choice for a patient. The general practitioner is not bound by a list of conditions.
9. Is there a list of doctors authorized to prescribe Medicinal Cannabis?
There is no list of authorized doctors who may prescribe Medicinal Cannabis as all general practitioners are allowed to do so. See further Question 8 of this Chapter.
10. What approvals or notifications are required to prescribe Medicinal Cannabis?
There are no specific approvals or notifications required to prescribe Medicinal Cannabis. See further Question 8 of this Chapter.
11. Where is Medicinal Cannabis available?
Medicinal Cannabis may be dispensed by pharmacies to patients. Moreover, a number of pharmacies in the Netherlands prepare oil from the BMC’s medicinal cannabis. Standardized cannabis oils are available on doctor’s prescription at certain pharmacies mentioned by the BMC on its website.
12. Is there a list of retailers authorized to sell Medicinal Cannabis?
No, there is no list of retailers authorized to sell medicinal cannabis.
13. Are there proposals for reform or significant change to the regulation of Medicinal Cannabis?
A review of the Dutch policy and regulation of medicinal cannabis is currently ongoing.
In a letter to Parliament dated May 2021, the Minister announced a possible revision of the role and tasks of the BMC. The Minister intends to reduce the BMC’s role in the trade in Medicinal Cannabis, including by reviewing the monopoly position and purchasing obligation of the BMC. To this end, the DOA will have to be amended. As of yet, it is unclear whether this will happen.
In a letter to Parliament dated March 2023, the Minister gave an update on the progress on the review of the Dutch medicinal cannabis policy. Firstly, the Minister expressed his content with the functioning of the closed production chain for medicinal cannabis under responsibility of the BMC so far (see further Question 4 of this Chapter). The Minister also proposed two policy adjustments to improve the opportunities within the current legal framework for medicinal cannabis.
- First, the Minister proposes removing the export ceiling and the restriction on production capacity which currently apply to Dutch exports of medicinal cannabis, to better meet international demand and provide more scope for international cooperation.
- Second, the Minister expresses his intention to review the framework for granting Opium Exemptions for parties that do not form part of the closed production chain for medicinal cannabis. He considers it desirable for these parties to be able to obtain an Opium Exemption if they are involved in the cultivation, processing or trading of medicinal cannabis and make a demonstrable contribution to scientific research into the medicinal application of cannabis and drug production (which serves the interests of the patient) and meet all the requirements set. It is currently not possible to buy medicinal cannabis from a supplier which is not contracted by the BMC. The Minister notes that this has an inhibiting effect on innovation, medical research and possibly also patient availability.
The Minister states that these policy amendments are a stepping stone towards future amendment of the DOA.
Opioid Drugs
1. Are Opioid Drugs authorized in your country?
Medicinal products with opioids as active substances
Similar to medicinal products with cannabinoids as active substances, medicinal products with opioids as active substances fall under the Medicines Act and are authorized in accordance with the general framework as discussed in Chapter 1, Question 3.
In addition, the prohibition regarding substances which are listed in List I (hard drugs) to that Act (“DOA List I Substances“) – including opioids – is not applicable in the case of activities within the normal professional practice of pharmacists and dispensing physicians to which the specific exemptions pursuant to Articles 4 and 5 DOA apply. The conditions for these exemptions are set out further in the DOA Decree. The DOA also provides that several designated institutes, such as licensed hospitals (listed in Article 16 DOA Decree), have a full exemption for the medicinal use of all List I and II substances. In other cases, an Opium Exemption for a specific prescription of List I Substances can be applied for (see further Cannabinoid Drugs, Question 1).
2. What are the regulatory authorities with jurisdiction over Opioid Drugs?
As mentioned in Question 1 of this Chapter, a distinction can be made between medicinal products with opioids as active substances and the medicinal use of DOA List I Substances. Since the regulatory framework applicable to medicinal products similarly applies to opioid medicinal products, please refer to Chapter 1, Question 1 of Exhibit A on the regulatory authorities.
For the medicinal use of opioids, the following regulatory authorities have jurisdiction:
Ministry of Health, Welfare and Sport
The Ministry of Health, Welfare and Sport, and specifically the Minister, is responsible for the granting of exemptions under the DOA and therefore responsible for the authorization of DOA List I Substances for the Dutch market.
Farmatec
Farmatec is mandated by the Minister for the granting of Opium Exemptions for DOA List I Substances on behalf of the Ministry of Health, Welfare and Sport.
The Inspectorate
The Inspectorate gives advice to Farmatec on applications received for an Opium Exemption. Moreover, the Inspectorate monitors the prescription and dispensing of DOA List I Substances to patients.
3. Is there a specific regulatory framework for the authorization, pricing, and reimbursement of Opioid Drugs?
Authorization
Please refer to Question 1 of this Chapter.
Pricing and reimbursement
There is no specific framework for pricing and reimbursement of opioid drugs. Health insurers will only reimburse a registered drug in as far as it is included in the Drug Reimbursement System. The medicinal products included in the Drug Reimbursement System are included in the annexes of the Healthcare Insurance Regulation (see further Exhibit A, Chapter 1, question 13). The Drug Reimbursement System includes opioid drugs; in particular, opioids provided for pain relief are covered by basic health insurance.
4. Which are the Opioid Drugs that have received market approval to date?
The ‘pharmacotherapeutic compass’ of the National Health Care Institute distinguishes medicinal products with opioids as active substances as a separate medicinal product group (please refer also to Chapter 8, question 4). A list of this product group can be
found on the Dutch language website of the National Health Care Institute here.
5. Who can prescribe Opioid Drugs?
Which persons may prescribe medicinal products, including those containing opioids as active substances, is regulated by the Individual Healthcare Professions Act; see further Chapter 1, Question 14.
DOA List I Substances may be prescribed by the healthcare professionals and designated institutes mentioned in the answer to Chapter 10, Question 1, provided that the conditions of the DOA Decree are complied with. The Decree contains (administrative) rules on the issuance, dispensing and storage of Opioid Drugs.
Furthermore, it follows from Article 2 of the DOA Decree that DOA List I Substances may under certain conditions be prescribed in the context of medical research within the meaning of the Medical Research (Human Subjects) Act.
6. Is there a list of doctors authorized to prescribe Opioid Drugs?
No, such a list does not exist; please refer to Chapter 1, Question 14, on the prescription of medicinal products.
Regarding the medicinal use of DOA List I Substances, a specific list of doctors authorized to prescribe DOA List I Substances does not exist either. In principle, all hospitals, pharmacists and general practitioners are exempt from the DOA. See also Question 1.
7. What approvals or notifications are required to prescribe Opioid Drugs?
Please refer to Chapter 1, Question 14.
8. Which organizations are authorized to sell/distribute Opioid Drugs available?
Please refer to Chapter 3, Question 18 regarding medicinal products with opioids as active substances.
Regarding the medicinal use of DOA List I Substances, please see Chapter 10, Question 1. Hospitals, pharmacists and general practitioners may be authorized to distribute DOA List I Substances.
9. Is there a list of retailers/distributors authorized to sell Opioid Drugs?
No, there is no list of retailers/distributers authorized to sell medicinal products with opioids as active substances. Neither is there a list of entities which may sell DOA List I Substances. In principle, selling DOA List I Substances is prohibited under the DOA. However, exceptions apply; see further Question 1 of this Chapter.
10. Are there proposals for reform or significant change to the regulation of Opioid Drugs?
A legislative proposal that entails adding a new list to the DOA (‘List IA’), which contains substance groups – instead of individual substances – whose chemical structure is derived from a number of DOA List I Substances, has been submitted to Parliament. The government thus proposes a generic ban on these substance groups. It is unclear if and when the bill will come into force. The original aim was to introduce it in the spring of 2022. However, at the time of writing, the proposal is yet to be discussed in Parliament. Moreover, in June 2022 the Council of State published advice on the subject in which it expressed doubts about the efficiency and effectiveness of the proposed bill.
11. When are they likely to come into force?
See Question 10 above.